 "
"
Team:Paris Bettencourt/Semantic containment
From 2012.igem.org

- Main
- Team
- Project
- Achievements
- Human Practice
- Safety
- Notebook
- Attributions
 Overview
Overview Delay system
Delay system Semantic containment
Semantic containment Restriction enzyme system
Restriction enzyme system MAGE
MAGE Suicide system
Suicide system Encapsulation
Encapsulation Synthetic import domain
Synthetic import domain Safety Questions
Safety Questions Safety Assessment
Safety AssessmentContents |
Overview
We want to prevent our genetic construct from conferring an advantage to other organisms. Horizontal gene transfer (HGT) can be performed either by conjugation, transduction, or transformation. As these processes involve two parties, our genetically modified bacteria and some wild type population, and as we will not modify wild type populations, we cannot assume that HGT is fully avoidable. Semantic containment [1] means that our bacteria won't be able to "speak" with other organisms, since they don't speak the same "language", the language being DNA. Our system will read the stop codon TAG as the amino-acid serine. It means that in our bacteria the stop codon will be translated into a serine, whereas in wild type bacteria this protein will be truncated and will not confer an advantage to these cells.

Semantic containment principle. The information carried by the DNA cannot be read by other organism
Objectives
Here we want to show that semantic containment works as expected. First we had to choose between two tRNA amber suppressor, either serine, or tyrosine. For that we calculate the abilities for the amber codon to reverse to a serine or tyrosine or related amino-acid that could conserve the function. Secondly we create a biobrick with a tRNA Amber suppressor (BBa_K914000), in order to have a reliable biobrick, with characterization of it. Thirdly, to test the latter biobrick, we built the biobrick BBa_K914009, that contain an an amber codon instead of one of it serine amino-acid.
With more time we will try to increase the robustness of this system, which is null when the tRNA amber suppressor is transferred too. We will try to create a new library of plasmid backbones in the part registry, where all backbones have at least two amber mutations. The idea is that all the community will be able to improve this library, either by adding new contained backbones, or by adding amber mutations on the same backbone.
Design
Calculation of serine and tyrosine weakness
In order to know which of these two amino-acids is the less robust to mutation from a 'TAG' codon (amber codon), we will calculate a score of weakness. The weakness of an amino-acid is defined here by its abilities to not revert to the same amino-acid or any other similar, from the amber codon. The score is calculated using the following formula :
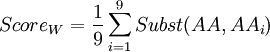
Where i is one of the nine amino-acids accessible after 1 mutation. Subst(AA,AAi) calculate the similarity score, using a BLOSUM62 matrix, between serine or tyrosine (AA) and one of the nine amino-acids around (AAi). The lowest score is the weakest.
Qualitative experiment : does it work?
We are going to transform a plasmid with a the Kan* gene into a MG1655 strain that contain either pSB1C3::supD, or pSB1C3::RFP. We plate them on Chloramphenicol and Kanamycin. The Kan* is supposed to be non functional without an amber suppressor.
Quantitative experiment : Is it working well? Is the amber mutation leaky?

We are going to use the following strains, all in the E. coli strain MG1655. To quantify the leakiness in terms of expression of the Kan* gene, we sill perform real time experiment, where we will measure the growth rate of each of these strains in different concentration of kanamycin. In case of leakiness, the Kan*+RFP strain will be able to grow in higher concentration of kanamycin than the negative control.
We will also be able to quantify the functionality of the Kan*+supD strain, by comparing with the positive control.
Experiments and results
Weakness calculation
We wrote a Python script that calculate the score of weakness, with all amino-acids (not only serine or tyrosine), and will sort a list of score, with the BLOSUM62, BLOSUM80, BLOSUM100.
The scores are, with a mutation rate of 10-9,and :
- BLOSUM62 :
- Serine : -1.0000000002222223e-09
- Tyrosine : 4.4444444307407385e-10
- BLOSUM80 :
- Serine : -1.444444445111111e-09
- Tyrosine : -2.222222242962964e-10
- BLOSUM100 :
- Serine : -3.333333334925926e-09
- Tyrosine : -1.0000000044444444e-09
So, no matter the matrix used, the amber codon is always less likely to revert into a serine or similar.
Also, we favor serine replacement over tyrosine, because the frequency of serine in the E coli genome is superior to the tyrosine one. It turns out that S has 57,88 codons over 1000 codons when Y has 28,59 codons over 1000 codons (Codon usage). Therefor, it might be more convenient to replace a serine than a tyrosine.
Qualitative characterization of K914000 and K914009

K914000 is the construction pLac-supD-T, and is named supD in the rest of the page. K914009 is the P1003 gene with the serine 133 which is replaced by a amber codon 'TAG'. Its name is Kan*.
After preparing electro-competent M1655 cells with either pSB1C3::supD or pSB1C3::RFP. I transform the plasmid pSB1A1::Kan* in both competent cells. After transformation, cells are plated on Cm+Kan.
We can observe that without any plasmids transformed no cells grow, or when we transform another plasmid with no Kan resistance gene, but with an Amp gene resistance (and plated on Cm+Amp), colonies appear in both strains. But the pSB1A1::Kan* can express the kanamycin resistance phenotype only in the strain containing the supD gene.
We can conclude here that the supD gene can rescue the phenotype KanS by allowing the correct expression of the kan gene P1003.
 MG1655+supD only |
 MG1655+RFP only |
 MG1655+supD+Kan* |
 MG1655+RFP+Kan* |
 MG1655+supD+pLac* |
Testing of the system
Experimental setup
Describe the experiment
Results
Present your results