 "
"
Team:Paris Bettencourt/Semantic containment
From 2012.igem.org
(→Overview) |
(→Calculation of serine and tyrosine weakness) |
||
| Line 35: | Line 35: | ||
</center> | </center> | ||
| - | Where i is one of the nine amino-acids accessible after 1 mutation. Subst(AA,AAi) calculate the similarity score, using a BLOSUM100 matrix, between serine or tyrosine (AA) and one of the nine amino-acids around (AAi). The lowest score is the weakest. The BLOSUM100 (BLOck SUbstitution Matrix) is constructed using local alignment of sequenced less than 100% identical<sup>[</sup><sup>[[# | + | Where i is one of the nine amino-acids accessible after 1 mutation. Subst(AA,AAi) calculate the similarity score, using a BLOSUM100 matrix, between serine or tyrosine (AA) and one of the nine amino-acids around (AAi). The lowest score is the weakest. The BLOSUM100 (BLOck SUbstitution Matrix) is constructed using local alignment of sequenced less than 100% identical<sup>[</sup><sup>[[#References|4,5]]</sup><sup>]</sup>, and is consequently adapted to appreciate the effect of a single mutation. |
===Qualitative experiment : does it work?=== | ===Qualitative experiment : does it work?=== | ||
Revision as of 10:52, 26 September 2012

- Main
- Team
- Project
- Achievements
- Human Practice
- Safety
- Notebook
- Attributions
 Overview
Overview Delay system
Delay system Semantic containment
Semantic containment Restriction enzyme system
Restriction enzyme system MAGE
MAGE Suicide system
Suicide system Encapsulation
Encapsulation Synthetic import domain
Synthetic import domain Safety Questions
Safety Questions Safety Assessment
Safety Assessment|
Achievements :
The first part (supD) is well characterized and works well. For the second parts, it turns out that this mutation is quite leaky, although it works in lab conditions, one mutation is not enough if we want to release such parts in nature. Other reasons emphasize this observation, notably the weakness of being at one mutation to recover the protein functionality.
|
Contents |
Overview
We want to prevent our genetic construct from conferring an advantage to other organisms. Horizontal gene transfer (HGT) can be performed either by conjugation, transduction, or transformation. As these processes involve two parties, our genetically modified bacteria and some wild type population, and as we will not modify wild type populations, we cannot assume that HGT is fully avoidable. Semantic containment[1] means that our bacteria won't be able to "speak" with other organisms, since they don't speak the same "language", the language being DNA. Our system will read the stop codon TAG as the amino-acid serine. It means that in our bacteria the stop codon will be translated into a serine, whereas in wild type bacteria this protein will be truncated and will not confer an advantage to these cells. The 'TAG' codon has been chosen because of its low frequency in E. coli genome (314 occurrences), and also because for further applications, Church Lab tries to remove all amber codon of an E. coli strain[2]. Although it has been demonstrate that the over-expression of a tRNA amber suppressor sole does not affect its growth rate nor the morphology of E. coli[3].

Objectives
Here we want to show that semantic containment works as expected. First we had to choose between two tRNA amber suppressor, either serine, or tyrosine. For that we calculate the abilities for the amber codon to reverse to a serine or tyrosine or related amino-acid that could conserve the function. Secondly we create a biobrick with a tRNA Amber suppressor ([http://partsregistry.org/Part:BBa_K914000 BBa_K914000]), in order to have a reliable biobrick, with characterization of it. Thirdly, to test the latter biobrick, we built the biobrick [http://partsregistry.org/Part:BBa_K914009 BBa_K914009], that contain an amber codon instead of one of it serine amino-acid. K914000 is the construction PLac-supD-T, and is named supD in the rest of the page. K914009 is the P1003 gene (kanamycin gene resistance) with the serine 133 which is replaced by a amber codon 'TAG'. Its name is Kan*.
With more time we will try to increase the robustness of this system, which is null when the tRNA amber suppressor is transferred too. We will try to create a new library of plasmid backbones in the part registry, where all backbones have at least two amber mutations. The idea is that all the community will be able to improve this library, either by adding new contained backbones, or by adding amber mutations on the same backbone.
Design
Calculation of serine and tyrosine weakness
In order to know which of these two amino-acids is the less robust to mutation from a 'TAG' codon (amber codon), we will calculate a score of weakness. The weakness of an amino-acid is defined here by its abilities to not revert to the same amino-acid or any other similar, from the amber codon. The score is calculated using the following formula :
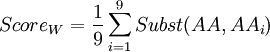
Where i is one of the nine amino-acids accessible after 1 mutation. Subst(AA,AAi) calculate the similarity score, using a BLOSUM100 matrix, between serine or tyrosine (AA) and one of the nine amino-acids around (AAi). The lowest score is the weakest. The BLOSUM100 (BLOck SUbstitution Matrix) is constructed using local alignment of sequenced less than 100% identical[4,5], and is consequently adapted to appreciate the effect of a single mutation.
Qualitative experiment : does it work?
To do so, we will transform a plasmid with a the Kan* gene into a MG1655 strain that contain either pSB1C3::supD, or pSB1C3::RFP. We plate them on Chloramphenicol and Kanamycin. The Kan* is supposed to be non functional without an amber suppressor.
Quantitative experiment : Is it working well? Is the amber mutation leaky?

We are going to use the following strains, all in the E. coli strain MG1655. To quantify the leakiness in terms of expression of the Kan* gene, we will perform real time experiment, where we will measure the growth rate (through OD600 measurement) of each of these strains in different concentrations of kanamycin. In case of leakiness, the Kan* + RFP strain (3) will be able to grow in higher concentration of kanamycin than the negative control (4).
We will also be able to quantify the functionality of the Kan*+supD strain, by comparing with the positive control.
Experiments and results
Weakness calculation
We wrote a Python script that can calculate the score of weakness, with all amino-acids (not only serine or tyrosine), and will sort a list of score. Data shown in appendix for other amino-acids.
The scores are, with a mutation rate of 10-9 for the serine and tyrosine:
- BLOSUM100 :
- Serine : ScoreSW = -3.33e-09
- Tyrosine : ScoreYW = -1.00e-09
The amber codon is less likely to revert into a serine or similar.
Also, we favor serine replacement over tyrosine, because the frequency of serine in the E. coli genome is superior to the tyrosine one. It turns out that S has 57,88 codons over 1000 codons when Y has 28,59 codons over 1000 codons ([http://openwetware.org/wiki/Escherichia_coli/Codon_usage Codon usage]). Therefor, it might be more convenient to replace a serine than a tyrosine, because proteins would more likely to contain many serines than tyrosines.
Qualitative characterization of K914000 and K914009

After preparing electro-competent M1655 cells with either pSB1C3::supD or pSB1C3::RFP. We transform the plasmid pSB1A1::Kan* in both competent cells. After transformation, cells are plated on Cm+Kan.
We can observe that without any plasmids transformed no cells grow, or when we transform another plasmid with no Kan resistance gene, but with an Amp gene resistance (and plated on Cm+Amp), colonies appear in the strain with RFP, unfortunately the other control (with supD) did not work this time, hence there is no picture of it. But for the quantitative experiment we need that control too (4), and we manage to do it that times. But the pSB1A1::Kan* can express the kanamycin resistance phenotype only in the strain containing the supD gene.
We can conclude here that the supD gene can rescue the phenotype KanS by allowing the correct expression of the kan gene P1003.
 MG1655+pSB1C3::supD only Selection : Cm+Amp |
 MG1655+pSB1C3::RFP only |
 MG1655+pSB1C3::supD+pSB1A1::Kan* |
 MG1655+pSB1C3::RFP+pSB1A1::Kan* |
 MG1655pSB1C3::+RFP+pSB1A2::pLac* |
Quantitative characterization of K914000 and K914009
Here we characterize both biobrick quantitatively. First, we are going to confirm the qualitative result for the part K914000 and then we will determine how a single amino-acid substitution is leaky.
Experimental setup
Once the construction is made (Fig. 2), we double transformed the plasmids into MG1655 strains, that does not contain any amber suppressor. We will work with three replicates of each strains, and for each strain we will be in 8 different conditions of antibiotic resistance. The antibiotic used is kanamycin, an aminoglycoside interfering with the translation. The range of concentration goes from 4 times as much as the usual concentration (100 µg/mL) to 8 times less. The 96 wells plate is then incubated in a plate reader that take measurements of OD600 every 6 minutes. This measure is correlated to the number of cell in the well. Each well contains 200µL of LB (Lysogeny broth, aka Luria Bertani), chloramphenicol and ampicilin at their usual concentration, the dilution of cells and different amount of kanamycin. An overlay of 50µL of mineral oil is added on the top. The measurement lasted approximately 16 hours and 30 min.

Results
First we observed that the qualitative result is reproduce here, the supD gene rescues the kanamycin resistance at any concentration of the antibiotic. It shows also that there is no disadvantage to use the supD amber suppressor compared to the wild type kanamycin gene resistance, since there is no difference of growth rate (Figure 4B and 4C). However the leakiness of the Kan* gene is higher than expected, and thus one mutation is not sufficient for the containment. Indeed, at usual concentration, Kan* gene manages to express an antibiotic resistance, even though lower than KanR or supD (Figure 4A). Here we define the growth rate as the OD600 observed at a given time (here t=8h20'), in order to overcome the fact that Kan*+RFP does not have a clear exponential phase. The fact that the strain (3) grows faster and can have higher OD600 may be explain by the fact that RFP can absorb at 584nm.


Conclusion & Perspectives
This work demonstrates that semantic containment can be achieved by changing an amino-acid into an amber codon. We saw that one mutation is not enough and further experiments should demonstrate that 2 mutations is much less leaky. The underlying idea is in fine to create a library of semantic-contained genes and backbones that would be improved by anybody, by either adding new semantic systems, or increasing the degree of containment by adding mutations.
Further experiments should improve the robustness of this system by adding a new security component to be at three horizontal genes transferred to fail. In our current system, the system fails if the tRNA amber suppressor gene is transferred with another semantic gene. The idea is to add a semantic containment for the tRNA. Since it is a tRNA we cannot replace amino-acids because it is not translated. In order to fix that problem we will construct the following system. The tRNA supD gene will be under a T7 promoter which is orthogonal, meaning that it needs a special RNA polymerase (T7 RNA polymerase) to transcribe the gene (here it would be supD), but here we would mutate the T7 RNA polymerase with several amber mutations. It means that we would need to transfer these two genes with the semantically contained gene in order to have something functional in the other organism, which is very unlikely to happen, further experiments will elucidate the probability of such an event. A schema of this system is depicted in Figure 5. This system needs to be activated in the lab, by transforming a plasmid with the wild type T7 RNA polymerase gene with another antibiotic gene resistance, say X. Then we remove antibiotic X and we will loose the plasmid with wild type T7 polymerase when the positive feedback loop has started.

References & Appendix
References
1 - Marliere, P. The farther, the safer : a manifesto for securely navigating synthetic species away from the old living world. System and Synthetic Biology 3, 77-84 (2009). [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2759432/ Paper]
2 - Isaacs, F.J. et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science (New York, N.Y.) 333, 348-53 (2011). [http://arep.med.harvard.edu/pdf/Isaacs_Sci_11.pdf Paper]
3 - Anderson, J.C., Voigt, C. a & Arkin, A.P. Environmental signal integration by a modular AND gate. Molecular systems biology 3, 133 (2007). [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1964800/ Paper]
4 - S Henikoff and J G Henikoff. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992 November 15; 89(22): 10915–10919. [http://www.ncbi.nlm.nih.gov/pmc/articles/PMC50453/pdf/pnas01096-0363.pdf Paper]
5 - Jorja G. Henikoff, Steven Henikoff, [6] Blocks database and its applications, Methods in Enzymology, Academic Press, 266, 88-105 (1996). [http://www.sciencedirect.com/science/article/pii/S007668799666008X Paper]
Appendix
Genotype of strain used
- MG1655 : F- λ- ilvG- rfb-50 rph-1
Other amino-acids weakness score
- Weakness score with a BLOSUM 100, mutation rate = 10-9, initial Codon : TAG
('A', [-4.000000002555556e-09])
('C', [-6.777777782148149e-09])
('D', [-5.2222222257407416e-09])
('E', [-3.222222225407407e-09])
('F', [-3.0000000040740746e-09])
('G', [-6.555555560444445e-09])
('H', [-2.888888891555555e-09])
('I', [-5.111111115777778e-09])
('K', [-2.6666666694074067e-09])
('L', [-4.333333337555555e-09])
('M', [-4.000000003185186e-09])
('N', [-4.3333333354074076e-09])
('P', [-5.888888893518519e-09])
('Q', [-1.6666666689629633e-09])
('R', [-3.888888891888888e-09])
('S', [-3.333333334925926e-09])
('T', [-4.22222222462963e-09])
('V', [-4.888888893111112e-09])
('W', [-2.4444444508518513e-09])
('Y', [-1.0000000044444444e-09])