 "
"
Team:TU-Delft/Modeling/StructuralModeling
From 2012.igem.org
MarkWeijers (Talk | contribs) |
|||
| (10 intermediate revisions not shown) | |||
| Line 6: | Line 6: | ||
<img src="https://static.igem.org/mediawiki/igem.org/7/74/Background_greenlamp.jpg" class="bg_team"> | <img src="https://static.igem.org/mediawiki/igem.org/7/74/Background_greenlamp.jpg" class="bg_team"> | ||
<div id="logo_ed"><a href="https://2012.igem.org/Team:TU-Delft" 'onfocus=this.blur()'><img src="https://static.igem.org/mediawiki/igem.org/e/e2/Igemlogo.png" border="0" width="100" height="100"></a></div> | <div id="logo_ed"><a href="https://2012.igem.org/Team:TU-Delft" 'onfocus=this.blur()'><img src="https://static.igem.org/mediawiki/igem.org/e/e2/Igemlogo.png" border="0" width="100" height="100"></a></div> | ||
| - | |||
| - | |||
| - | |||
| - | |||
<div id="contentbox" style="text-align:justify;"> | <div id="contentbox" style="text-align:justify;"> | ||
| + | |||
| + | <img src="https://static.igem.org/mediawiki/igem.org/7/77/Structuralmodeling.jpg" align="middle" width="100%"> | ||
| + | |||
<h2>Summary</h2> | <h2>Summary</h2> | ||
| Line 103: | Line 102: | ||
<img src="https://static.igem.org/mediawiki/2012/b/be/Table2_energy.png" name="kugroup" width="570" border="0" id="kugroup" /></a> | <img src="https://static.igem.org/mediawiki/2012/b/be/Table2_energy.png" name="kugroup" width="570" border="0" id="kugroup" /></a> | ||
<br/> | <br/> | ||
| - | <h6>[a] Calculated binding energy by YASARA. | + | <h6>TMH1 stands for TransMembrane Helix 1, ECL2 stands for ExtraCellular Loop 2.</h6> |
| - | [b] Two criteria for receptor activation; temporary bonding to Ser 247 (> 25%) and fluctuating bonding to Arg 111 (<0%), and 1) Robust bonding to Ser 178 and Arg 251 (>50%) and 2) fluctuating and temporary bonding to Ser 178 (0-24%) and Arg 251 (25-49%) respectively with temporary bonding between Asn 45 and Ser 287. | + | <h6>[a] Calculated binding energy by YASARA.</h6> |
| - | [c] <i>In vivo</i> activity qualified as EC50 response <20, otherwise inactive. | + | <h6>[b] Two criteria for receptor activation; temporary bonding to Ser 247 (> 25%) and fluctuating bonding to Arg 111 (<0%), and 1) Robust bonding to Ser 178 and Arg 251 (>50%) and 2) fluctuating and temporary bonding to Ser 178 (0-24%) and Arg 251 (25-49%) respectively with temporary or robust bonding between Asn 45 and Ser 287.</h6> |
| - | [d] Mutated residue variant compared to its relevant residue. | + | <h6>[c] <i>In vivo</i> activity qualified as EC50 response <20, otherwise inactive. EC50 data from Offermans et al.[4]</h6> |
| - | [e] NR1 receptor with ligand methyl nicotinate. </h6> | + | <h6>[d] Mutated residue variant compared to its relevant residue.</h6> |
| + | <h6>[e] NR1 receptor with ligand methyl nicotinate. </h6> | ||
<br/> | <br/> | ||
| - | <p>Table 1 shows the percentage of the total simulation time at which the amino acids are bound to the ligand. Each MD simulation was executed after an energy minimization and the specific point-mutation during a simulation time of 1 nanosecond. The active/inactive states are partly based on the classification of the responses of the hOR2AG1 mutants; every EC50 value of 20 or higher is classified as being inactive and having a low affinity. Every response below 20 is considered active.</p> | + | <p>Table 1 shows the percentage of the total simulation time at which the amino acids are bound to the ligand. Each MD simulation was executed after an energy minimization and the specific point-mutation during a simulation time of 1 nanosecond. The active/inactive states are partly based on the classification of the responses of the hOR2AG1 mutants; every EC50 value (from the research of Offermanns et al.[4]) of 20 or higher is classified as being inactive and having a low affinity. Every response below 20 is considered active.</p> |
<p>In addition, in the paper hydrogen-bonds in time are also classified in groups; whenever there is a hydrogen-bond present for 1-24% of the simulation time, it is considered temporary bonds, fluctuating during 25-49% of the time, and robust bonds are considered when they are bound 50-100% of the simulation time. </p> | <p>In addition, in the paper hydrogen-bonds in time are also classified in groups; whenever there is a hydrogen-bond present for 1-24% of the simulation time, it is considered temporary bonds, fluctuating during 25-49% of the time, and robust bonds are considered when they are bound 50-100% of the simulation time. </p> | ||
| - | <p>Analyses of table 1 shows primarily that for a hGPR109A mutant to be active, at least the protein-ligand interaction for amino acid Ser 178 and Arg 251 have to be >50, together with Ser 247 and Arg 111 , which should have a value of >25 and >0, respectively. However, this is not the case for mutant L83V, which has a very low value for its hydrogen-bonding to Ser 178 and Arg 251. When checking for the internal hydrogen-bonds, one in particular stood in correlation with this mutant and the others. The hydrogen-bonding of Asn 45 to Ser 287 can be considered robust in comparison with the other active mutants, which are <50. </p> | + | <p>Analyses of table 1 shows primarily that for a hGPR109A mutant to be active, at least the protein-ligand interaction for amino acid Ser 178 and Arg 251 have to be >50, together with Ser 247 and Arg 111 , which should have a value of >25 and >0, respectively. However, this is not the case for mutant L83V, which has a very low value for its hydrogen-bonding to Ser 178 and Arg 251. When checking for the internal hydrogen-bonds, one in particular stood in correlation with this mutant and the others. The hydrogen-bonding of Asn 45 to Ser 287 can be considered robust in comparison with the other active mutants, which are <50. This means that this bonding between transmembrane helix 1 and 7 is a sort of "fail-safe" mechanism that comes into action when both hydrogen-bondings of Ser 178 and Arg 251 to the ligand fail.</p> |
<p>The inactive mutants all have a value of <50 for Arg 251 which classifies them as being inactive, except for N86Y and R111A. The latter mutant has no Arg 111 at all, and can be considered as being inactive by definition. The hydrogen-bonding of Arg 251 to the ligand is robust, however the protein-ligand interaction of Ser 178 very low. In comparison to the data of the mutant L83V, for the mutant N86Y to be inactive, the interaction between Ser 287 and Asn 45 should be <50, which is the case. </p> | <p>The inactive mutants all have a value of <50 for Arg 251 which classifies them as being inactive, except for N86Y and R111A. The latter mutant has no Arg 111 at all, and can be considered as being inactive by definition. The hydrogen-bonding of Arg 251 to the ligand is robust, however the protein-ligand interaction of Ser 178 very low. In comparison to the data of the mutant L83V, for the mutant N86Y to be inactive, the interaction between Ser 287 and Asn 45 should be <50, which is the case. </p> | ||
| Line 125: | Line 125: | ||
<br/> | <br/> | ||
| - | <p> Molecular Dynamics analyses of the NR1-receptor shows a result similar to mutant N86Y. However, in this case the Ser 187 - Asn 45 bonding (which is also present in the NR1 receptor) is active for 38% of the time. The boundary condition for this interaction to have influence on a weak protein-ligand bonding of Ser 178 or Arg 251, could be fixed at >25%. The interaction of methyl nicotinate to the NR1-receptor clearly shows no affinity at all, which can also be concluded from the dramatic drop in binding energy.</p> | + | <p> Molecular Dynamics analyses of the NR1-receptor (see movie) shows a result similar to mutant N86Y. However, in this case the Ser 187 - Asn 45 bonding (which is also present in the NR1 receptor) is active for 38% of the time. The boundary condition for this interaction to have influence on a weak protein-ligand bonding of Ser 178 or Arg 251, could be fixed at >25%, instead of >50% as mentioned earlier. If this is because the NR1-receptor has different properties than the GPR109A on the overal scale of the receptor, further research should figure out. The interaction of methyl nicotinate to the NR1-receptor clearly shows no affinity at all, which can also be concluded from the dramatic drop in binding energy.</p> |
<br/> | <br/> | ||
<br/> | <br/> | ||
| Line 132: | Line 132: | ||
<br/> | <br/> | ||
| - | <p>Furthermore, another interaction of Arg 251 with its neighboring amino acid, Glu 196, was investigated. This hydrogen-bond can be considered as being very robust, whereas the bonding is always 99% or higher, except of course for mutant R251A. It seems that this mutant may have a role to play in the conformational change of the receptor, but in the absence of the amino acid the receptor is still able to be activated, but many times less than the non-mutated wildtype receptor. From this can be suggested that Arg 251 indeed may be mutated in the | + | <p>Furthermore, another interaction of Arg 251 with its neighboring amino acid, Glu 196, was investigated. This hydrogen-bond can be considered as being very robust, whereas the bonding is always 99% or higher, except of course for mutant R251A. It seems that this mutant may have a role to play in the conformational change of the receptor, but in the absence of the amino acid the receptor is still able to be activated, but many times less than the non-mutated wildtype receptor. From this can be suggested that Arg 251 indeed may be mutated in the reengineering assay, if further investigation turns out that the bonding between Ser 287 and Asn 45 does not play a large role in compensating the absense of Arg 251. </p> |
<br/> | <br/> | ||
| Line 167: | Line 167: | ||
<br/> | <br/> | ||
| - | <h6> | + | <h6>[1] Syhre M, Chambers ST (2008) The scent of Mycobacterium tuberculosis. Tuberculosis. 88:317–323</h6> |
| - | [1] Syhre M, Chambers ST (2008) The scent of Mycobacterium tuberculosis. Tuberculosis. 88:317–323 | + | <h6>[2] Gelis L, Wolf S, Hatt H, Neuhaus EM, Gerwert K (2012) Prediction of a Ligand-Binding Niche within a Human Olfactory Receptor by Combining Site-Directed Mutagenesis with Dynamic Homology Modeling. Angew. Chem. Int. Ed. 51:1274-1278</h6> |
| - | [2] Gelis L, Wolf S, Hatt H, Neuhaus EM, Gerwert K (2012) Prediction of a Ligand-Binding Niche within a Human Olfactory Receptor by Combining Site-Directed Mutagenesis with Dynamic Homology Modeling. Angew. Chem. Int. Ed. 51:1274-1278 | + | <h6>[3] Jasmina Minic, Marie-annick Persuy, Elodie Godel, Josiane Aioun, Ian Connerton, Roland Salesse, Functional expression of olfactory receptors in yeast and development of a bioassay for odorant screening, FEBS Journal (2005)</h6> |
| - | [3] Jasmina Minic, Marie-annick Persuy, Elodie Godel, Josiane Aioun, Ian Connerton, Roland Salesse, Functional expression of olfactory receptors in yeast and development of a bioassay for odorant screening, FEBS Journal (2005) | + | <h6>[4] Tunaru S, Lättig J, Kero J, Krause G, Offermanns S (2005) Characterization of Determinants of Ligand Binding to the Nicotinic Acid Receptor GPR109A (HM74A/PUMA-G). Mol Pharmacol. 68:1271-1280</h6> |
| - | [4] Tunaru S, Lättig J, Kero J, Krause G, Offermanns S (2005) Characterization of Determinants of Ligand Binding to the Nicotinic Acid Receptor GPR109A (HM74A/PUMA-G). Mol Pharmacol. 68:1271-1280 | + | <h6>[5]J Zhang, Y Zhang. GPCR-ITASSER: A new composite algorithm for G protein-coupled receptor structure prediction and the application on human genome. 2011</h6> |
| - | [5]J Zhang, Y Zhang. GPCR-ITASSER: A new composite algorithm for G protein-coupled receptor structure prediction and the application on human genome. 2011</h6> | + | |
Latest revision as of 04:02, 27 October 2012



Summary
In order to engineer a yeast strain that is able to detect a tuberculosis (TB) molecule like methyl nicotinate, its receptor should be designed in such a way that the molecule can act as an agonist. By modeling the olfactory receptor in silico, its biophysical and biochemical properties are investigated at the molecular level. The aim is to get a clear understanding of how a ligand binds in the receptor and how to mutate the binding niche to let it bind to methyl nicotinate more specifically.
A validated model of the human niacin receptor 1 was built and a prediction criteria was derived from experimental data of mutated variants of this receptor. Also, the human OR2AG1 receptor was successfully reproduced with its ligand docked inside. An additional hydrogen-bonding of two amino acids within the receptor was found to have an considerable influence on the conformational change properties.
It is expected that this prediction method could give more reliable models of odorant receptors and thus engineer many receptors for ligands like fruit odors, compounds exhaled by people with a disease, sense explosives and drugs.
Introduction
Within the Snifferomyces project, the team of iGEM TU Delft 2012 aims to develop an olfactory device to sense volatile compounds. As an application, a few snifferomyces receptors were engineered in yeast to detect molecules in the breath of a tuberculosis (TB) patient (Figure 1a-d) and to detect banana smell.

Figure 1. Chemical compounds present in the breath of a TB-patient, represented both in 2D as in 3D.[1] Methyl nicotinate (a), methyl phenylacetate (b), methyl p-anisate (c) and o-phenylanisole (d)
These included the niacin receptor 1 of Rattus Norvegicus (a.k.a. rGPR109A) and the banana receptor of Mus Musculus (a.k.a. mOlfr154). By replacing the first alpha-helix, N- and C-terminals by the ones of the I7 receptor of Rattus Norvegicus, the receptor is known to be integrated in the cell membrane [3]. These two Snifferomyces receptors, niacin receptor (NR1) and banana receptor (BR4), together with their I7 flanks, were to be modeled in molecular dynamics software, named YASARA. This way, a prediction method of the receptor activity could be implemented for use on both these receptors. However, the NR1-receptor is not specifically evolved to detect one of the TB-molecules, methyl nicotinate (Figure 1a), but niacin. In theory, the BR4-receptor can be reengineered in such a way that it could let the second TB-molecule, methyl phenylacetate (Figure 1b) act as an agonist.
Prediction method
The research group of Gerwert et al. [2] developed a method based on Molecular Dynamics (MD) simulations and experimental data, to predicted whether a molecule would act as an agonist for the hOR2AG1 receptor. Within their research, point-mutations were inserted in the gene of the hOR2AG1 receptor, in order to lower the activity hardly or dramatically. By incorporating these point-mutations in their model, they could predict whether such a mutation would indeed influence the activity of the receptor. They concluded that if a molecule would be hydrogen-bonded for >50% of 10 nanoseconds simulation time to amino acid Thr 279, and > 0% of the same time period to Ser 263 or Ser 264 in the binding niche, the receptor would be activated. This requirement was specifically targeted for molecules with an ester-configuration, just like amyl butyrate and isoamyl acetate are.
To examine whether a similar requirement could be acquired from the NR1-model, a similar analysis approach was developed by using the experimental data from the hGPR109A mutants [4]. The EC50 responses of these mutants were normalized to the response of the wildtype (WT) hOR2AG1 receptor. Some mutants were simulated together with niacin docked in their binding niche, by using Molecular Dynamics in YASARA. Instead of 10 ns simulation time, 1 ns was chosen as simulation time, mainly due to the fact that a simulation of 10 ns would take 7-8 days to complete. The results of these simulations are analyzed on hydrogen-bonding between amino acids and the ligand. That information was generated for all mutants and the WT, and thus compared to one another, to see whether there is a correlation in the hydrogen-bonded protein-ligand interaction.
In addition, all other internal hydrogen-bonds between the amino acids of the receptor were analyzed to examine whether a point-mutation has influence on these hydrogen-bonds as well, even if the point-mutation did not disrupt the binding niche directly. It was proposed that a disruption in the hydrogen-bonding between several amino acids might influence the conformational change of the receptor, thus may not activate the receptor and in turn not the G-alpha protein. Analyses of the MD results on the hGPR109A and hOR2AG1 should give an insight whether other hydrogen-bonds then the ones bound to the ligand influence the conformational change.
Reengineering of binding niche
One of the main objectives the Snifferomyces project consisted of, was to engineer a receptor that has an affinity to methyl nicotinate. Because this compound and niacin are very close related, the choose to use this receptor as an template was obvious. In order to let this receptor sense methyl nicotinate with a high affinity, the binding-niche should be reprogrammed in order to let it bind. With the information on the internal hydrogen-bonding, several residues surrounding the ligand can be mutated without causing an disruption in the conformational change.
The same may hold for the BR4 receptor, which is a receptor to the ester amyl butyrate (apricot smell). The second TB-molecule, methyl phenylacetate, is also an ester. MD simulations and experimental data on the hOR2AG1 receptor [2] showed that the affinity on the ligand isoamylbenzoate (papaya smell) was increased after one point-mutation. MD simulations on the BR4-receptor with the second TB-molecule, methyl phenylacetate, should indicate whether a point-mutation for higher affinity should be necessary.
Results
BR4 Model
The BR4-model was based upon the human OR2AG1 (a.k.a. hOR2AG1) receptor, which has amyl butyrate [FIG] as an agonist. Previous research had modeled the hOR2AG1 receptor in silico and by using a homology modeling macro of YASARA, this model was reproduced conform the alignment done by Gerwert et.al.[2]

Figure 2. Reproduction of the hOR2AG1 binding-niche [2] by homology modeling.
After aligning the hOR2AG1 receptor with the BR4 receptor, and using the same swapping method as described for the NR1 model, the BR4-model was constructed. However, time did not permit us to execute simulations on the hOR2AG1 to test if the model would hold the same binding-niche characteristics. As soon as this model is conformed to the characteristics of the model from the paper, the BR4 sequence can be aligned with it to build the model.
NR1 Model
The model of the NR1 receptor was based upon the predicted model of Zhang I-Tasser website [5]. In turn, this model of the human niacin receptor 1 (a.k.a. hGPR109A) was predicted by the Zhang research group. By aligning the construct of our NR1 amino acid sequence with hGPR109A (Figure 3), a homology model was built by swapping the different amino acids of hGPR109A by the ones of NR1.

Figure 3. Alignment of hGPR109A and Snifferomyces receptor NR1.
Several hGPR109A mutants from the data of Offermanns et al. [4] were chosen to be used in the MD simulations, which can be seen in table 1.
Table 1. In silico hydrogen-bond contacts of niacin with Homo sapiens GPR109A variants. The percentages represent frequency of hydrogen-bond contact occurrence.
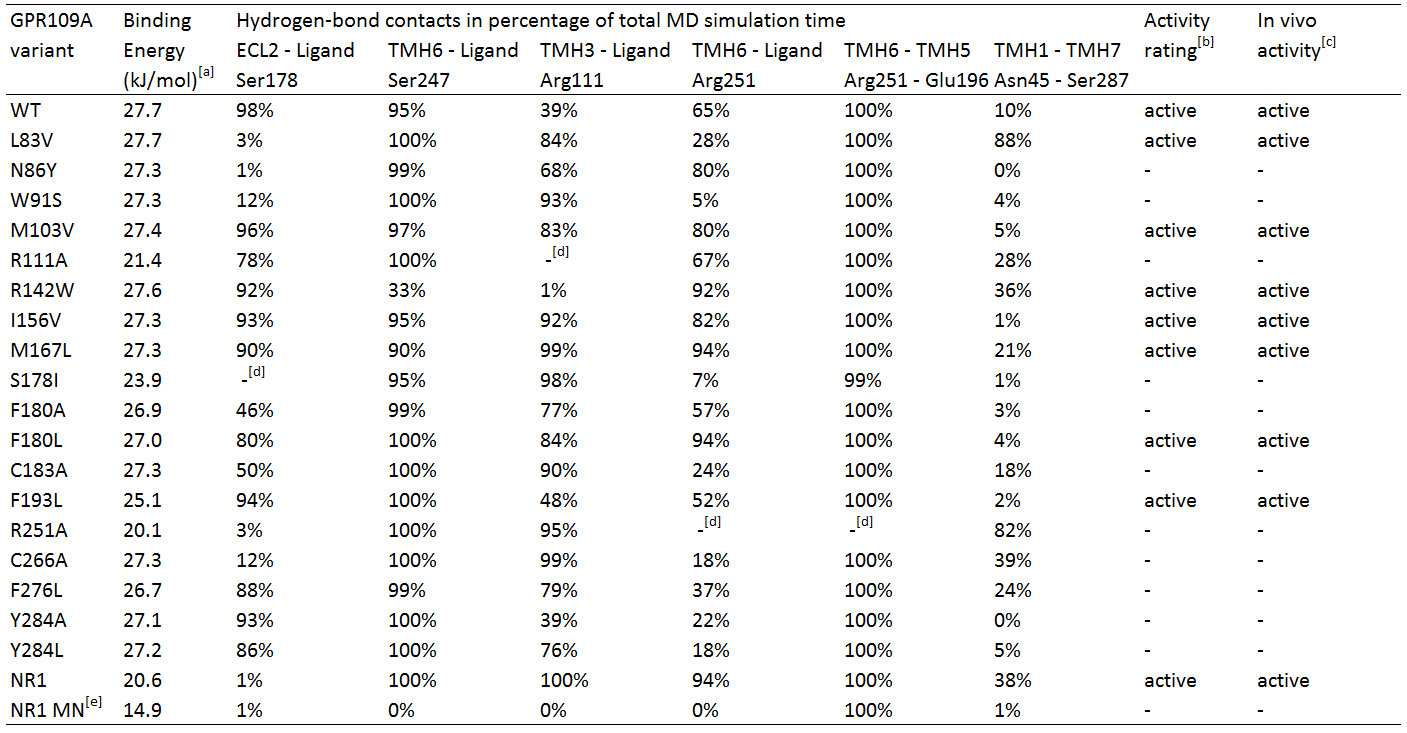
TMH1 stands for TransMembrane Helix 1, ECL2 stands for ExtraCellular Loop 2.
[a] Calculated binding energy by YASARA.
[b] Two criteria for receptor activation; temporary bonding to Ser 247 (> 25%) and fluctuating bonding to Arg 111 (<0%), and 1) Robust bonding to Ser 178 and Arg 251 (>50%) and 2) fluctuating and temporary bonding to Ser 178 (0-24%) and Arg 251 (25-49%) respectively with temporary or robust bonding between Asn 45 and Ser 287.
[c] In vivo activity qualified as EC50 response <20, otherwise inactive. EC50 data from Offermans et al.[4]
[d] Mutated residue variant compared to its relevant residue.
[e] NR1 receptor with ligand methyl nicotinate.
Table 1 shows the percentage of the total simulation time at which the amino acids are bound to the ligand. Each MD simulation was executed after an energy minimization and the specific point-mutation during a simulation time of 1 nanosecond. The active/inactive states are partly based on the classification of the responses of the hOR2AG1 mutants; every EC50 value (from the research of Offermanns et al.[4]) of 20 or higher is classified as being inactive and having a low affinity. Every response below 20 is considered active.
In addition, in the paper hydrogen-bonds in time are also classified in groups; whenever there is a hydrogen-bond present for 1-24% of the simulation time, it is considered temporary bonds, fluctuating during 25-49% of the time, and robust bonds are considered when they are bound 50-100% of the simulation time.
Analyses of table 1 shows primarily that for a hGPR109A mutant to be active, at least the protein-ligand interaction for amino acid Ser 178 and Arg 251 have to be >50, together with Ser 247 and Arg 111 , which should have a value of >25 and >0, respectively. However, this is not the case for mutant L83V, which has a very low value for its hydrogen-bonding to Ser 178 and Arg 251. When checking for the internal hydrogen-bonds, one in particular stood in correlation with this mutant and the others. The hydrogen-bonding of Asn 45 to Ser 287 can be considered robust in comparison with the other active mutants, which are <50. This means that this bonding between transmembrane helix 1 and 7 is a sort of "fail-safe" mechanism that comes into action when both hydrogen-bondings of Ser 178 and Arg 251 to the ligand fail.
The inactive mutants all have a value of <50 for Arg 251 which classifies them as being inactive, except for N86Y and R111A. The latter mutant has no Arg 111 at all, and can be considered as being inactive by definition. The hydrogen-bonding of Arg 251 to the ligand is robust, however the protein-ligand interaction of Ser 178 very low. In comparison to the data of the mutant L83V, for the mutant N86Y to be inactive, the interaction between Ser 287 and Asn 45 should be <50, which is the case.
Choose 720p HD for the best visualization. In case the place seems to be blank, zoom into the page by using Ctrl + Mouse-wheel and hit F5.
Molecular Dynamics analyses of the NR1-receptor (see movie) shows a result similar to mutant N86Y. However, in this case the Ser 187 - Asn 45 bonding (which is also present in the NR1 receptor) is active for 38% of the time. The boundary condition for this interaction to have influence on a weak protein-ligand bonding of Ser 178 or Arg 251, could be fixed at >25%, instead of >50% as mentioned earlier. If this is because the NR1-receptor has different properties than the GPR109A on the overal scale of the receptor, further research should figure out. The interaction of methyl nicotinate to the NR1-receptor clearly shows no affinity at all, which can also be concluded from the dramatic drop in binding energy.
Reengineering of the NR1 receptor
Furthermore, another interaction of Arg 251 with its neighboring amino acid, Glu 196, was investigated. This hydrogen-bond can be considered as being very robust, whereas the bonding is always 99% or higher, except of course for mutant R251A. It seems that this mutant may have a role to play in the conformational change of the receptor, but in the absence of the amino acid the receptor is still able to be activated, but many times less than the non-mutated wildtype receptor. From this can be suggested that Arg 251 indeed may be mutated in the reengineering assay, if further investigation turns out that the bonding between Ser 287 and Asn 45 does not play a large role in compensating the absense of Arg 251.
Discussion
While the method of this research was mainly based upon the experimental and MD data from Gerwert et.al.[2], not all MD requirements as done by them were taken into account. The main differences were the use of MD simulation program (instead of GROMACS, YASARA was used) and the simulation time was a 10-fold lower. The higher the simulation time, the more the simulation time is similar to the real protein-ligand interaction time.
One should also take in account that the models of the hGPR109A and hOR2AG1 receptors are an approximation of reality. By doing point-mutations on these receptors, the models can be refined. For this research only a limited amount of hGPR109A mutants were available. For example, a point-mutation in Ser 247 would give more insight on the affinity of the binding niche. In addition, the NR1 and BR4 models are derivatives of the hGPR109A and hOR2AG1, respectively. The alignments included no gaps, which means that the swapping method could give a reliable representation of the Snifferomyces receptors.
To have a more accurate model of the NR1 and BR4 receptors, point-mutations on these receptors and measurement of the response to certain ligands should be implemented in further research. By running MD simulations, interesting amino acids can be selected for mutation to examine whether the expected response is generated by the receptor.
Point-mutations on possible important amino acids like Asn 45 and Ser 287 should give an understanding whether the interaction between these two residues really act as a “fail-safe” mechanism that could still activate the G-alpha protein. Further research could give more insight into other possible residues that could be responsible for such a mechanism.
The simulation times were a crucial part of the structural modeling in YASARA. YASARA has a friendly user's interface and has many integrated features. However, the MD simulations are optimized for use on a 8-core computer. This means that a simulation of 10 ns takes about 7-8 days to completely simulate on a 16-core computer, let alone an 8-core computer. Also, by taking in account the error probability for every simulation, this process exhausts a lot of time.
Thankfully, the SARA institute was willing to help us and assist us in setting up the environment to use their HPC Cloud Server remotely from our office at Delft University. This gave us the opportunity to execute our simulations on a configurable amount of cores. By testing what amount of cores was the fastest to use (but not necessarily the most efficient) short simulations on 4, 8, 12, 16, 18, 20 and 24 cores were performed. The outcome was in favor of the 16-core computer. This means that a simulation of 10 ns would take 7-8 days. In contrast, a simulation of 1 nanosecond took about 2-2.5 days.
.Follow-up
Some additional ideas on structural modeling had to be given a lower priority. An interesting approach would be to engineer an olfactory receptor for one of the other three chemical compounds (Figure 1b-d) which are found in the breath of a tuberculosis patient. In the same manner, the reprogramming of the binding niche could allow different derivatives of a certain compound (methyl isonicotinate instead of methyl nicotinate) allow to bind, of which a kind of universality could be investigated to predict a configuration of the binding niche to any ligand.
Another follow-up would be to investigate in silico how large the conformational change of the receptor is, and see how this correlates to the hydrogen bonding, RSMD and energy. This would require data on simulations done with and without a ligand. This could eventually lead to a value for the dissociation rate between the G-alpha protein and the receptor.
Because the hOR2AG1 is a receptor to a fruity molecule, there is a potential that it could sense other odors van fruits that hold an ester-configuration as well. Many of these odors are derivatives of each other, so the BR4 receptor could be reengineered in such a way that it can sense any fruity molecule.
References
[1] Syhre M, Chambers ST (2008) The scent of Mycobacterium tuberculosis. Tuberculosis. 88:317–323
[2] Gelis L, Wolf S, Hatt H, Neuhaus EM, Gerwert K (2012) Prediction of a Ligand-Binding Niche within a Human Olfactory Receptor by Combining Site-Directed Mutagenesis with Dynamic Homology Modeling. Angew. Chem. Int. Ed. 51:1274-1278
[3] Jasmina Minic, Marie-annick Persuy, Elodie Godel, Josiane Aioun, Ian Connerton, Roland Salesse, Functional expression of olfactory receptors in yeast and development of a bioassay for odorant screening, FEBS Journal (2005)
[4] Tunaru S, Lättig J, Kero J, Krause G, Offermanns S (2005) Characterization of Determinants of Ligand Binding to the Nicotinic Acid Receptor GPR109A (HM74A/PUMA-G). Mol Pharmacol. 68:1271-1280
[5]J Zhang, Y Zhang. GPCR-ITASSER: A new composite algorithm for G protein-coupled receptor structure prediction and the application on human genome. 2011
