 "
"
Team:BostonU/Notebook
From 2012.igem.org
| (133 intermediate revisions not shown) | |||
| Line 41: | Line 41: | ||
color: #B22222; | color: #B22222; | ||
font-size: 28pt; | font-size: 28pt; | ||
| + | } | ||
| + | |||
| + | H4 { | ||
| + | font-family: Signika; | ||
| + | text-transform: uppercase; | ||
| + | text-decoration: none; | ||
| + | text-align: left; | ||
| + | color: #B22222; | ||
| + | font-size: 20pt; | ||
} | } | ||
| Line 49: | Line 58: | ||
text-align: left; | text-align: left; | ||
color: #696969; | color: #696969; | ||
| - | font-size: | + | font-size: 12pt; |
} | } | ||
| - | + | H9 { | |
font-family: Electrolize; | font-family: Electrolize; | ||
text-transform: none; | text-transform: none; | ||
| Line 59: | Line 68: | ||
color: #B22222; | color: #B22222; | ||
font-size: 12pt; | font-size: 12pt; | ||
| + | } | ||
| + | |||
| + | H7 { | ||
| + | font-family: Signika; | ||
| + | text-transform: none; | ||
| + | text-decoration: none; | ||
| + | text-align: left; | ||
| + | color: #000000; | ||
| + | font-size: 11pt; | ||
| + | } | ||
| + | H8 { | ||
| + | font-family: Signika; | ||
| + | text-transform: none; | ||
| + | text-decoration: none; | ||
| + | text-align: left; | ||
| + | color: #B22222; | ||
| + | font-size: 14pt; | ||
| + | } | ||
| + | |||
| + | H2 { | ||
| + | font-family: Signika; | ||
| + | text-transform: none; | ||
| + | text-decoration: none; | ||
| + | text-align: left; | ||
| + | color: #B22222; | ||
| + | font-size: 14pt; | ||
} | } | ||
</style> | </style> | ||
| - | <link rel="stylesheet" type="text/css" href="http://static.tumblr.com/bepcnvc/ | + | <link rel="stylesheet" type="text/css" href="http://static.tumblr.com/bepcnvc/zlPm6ibrh/team.css"> |
<link href='http://fonts.googleapis.com/css?family=Signika' rel='stylesheet' type='text/css'> | <link href='http://fonts.googleapis.com/css?family=Signika' rel='stylesheet' type='text/css'> | ||
<link href='http://fonts.googleapis.com/css?family=Electrolize' rel='stylesheet' type='text/css'> | <link href='http://fonts.googleapis.com/css?family=Electrolize' rel='stylesheet' type='text/css'> | ||
| Line 72: | Line 107: | ||
<div id="bu-wellesley_wiki_content"> | <div id="bu-wellesley_wiki_content"> | ||
| - | <p style="text-align:center;"><a href="https://2012.igem.org/Team:BostonU"><img src=" | + | <p style="text-align:center;"><a href="https://2012.igem.org/Team:BostonU"><img src="https://static.igem.org/mediawiki/2012/2/2d/NotebookBU.png" width="800px"></a></p> |
<ul id="nav"> | <ul id="nav"> | ||
| Line 84: | Line 119: | ||
<ul> | <ul> | ||
<li><a href="https://2012.igem.org/Team:BostonU/Project_Overview">Project Overview and Abstract</a></li> | <li><a href="https://2012.igem.org/Team:BostonU/Project_Overview">Project Overview and Abstract</a></li> | ||
| - | <li><a href="https://2012.igem.org/Team:BostonU/Characterization">Introduction to Characterization</a></li> | + | <li><a href="https://2012.igem.org/Team:BostonU/MoClo2">Introduction to MoClo</a></li> |
| + | <li><a href="https://2012.igem.org/Team:BostonU/Characterization">Introduction to Characterization</a></li> | ||
| + | <li><a href="https://2012.igem.org/Team:BostonU/DataSheet">Introduction to Data Sheets</a></li> | ||
<li><a href="https://2012.igem.org/Team:BostonU/Methodology ">Methodology Overview</a></li> | <li><a href="https://2012.igem.org/Team:BostonU/Methodology ">Methodology Overview</a></li> | ||
<li><a href="https://2012.igem.org/Team:BostonU/Results ">Results Summary</a></li> | <li><a href="https://2012.igem.org/Team:BostonU/Results ">Results Summary</a></li> | ||
| Line 110: | Line 147: | ||
<li><a href="#">Considerations</a> | <li><a href="#">Considerations</a> | ||
<ul> | <ul> | ||
| - | <li><a href="https://2012.igem.org/Team:BostonU/ | + | <li><a href="https://2012.igem.org/Team:BostonU/NEGEM">New England iGEM Regional Meeting</a></li> |
<li><a href="https://2012.igem.org/Team:BostonU/Human Practices">Human Practices</a></li> | <li><a href="https://2012.igem.org/Team:BostonU/Human Practices">Human Practices</a></li> | ||
<li><a href="https://2012.igem.org/Team:BostonU/Safety">Safety</a></li> | <li><a href="https://2012.igem.org/Team:BostonU/Safety">Safety</a></li> | ||
| Line 127: | Line 164: | ||
<br> | <br> | ||
| - | < | + | <h4>Weekly Notebook</h4> |
<br> | <br> | ||
| - | < | + | |
| + | <div id="tracking_nav"> | ||
| + | JUMP TO...<br> | ||
| + | <a href="#bu-wellesley_wiki_content">Top</a><br> | ||
| + | <a href="#May5">May Week 5</a><br> | ||
| + | <a href="#June1">June Week 1</a><br> | ||
| + | <a href="#June2">June Week 2</a><br> | ||
| + | <a href="#June3">June Week 3</a><br> | ||
| + | <a href="#June4">June Week 4</a><br> | ||
| + | <a href="#July1">July Week 1</a><br> | ||
| + | <a href="#July2">July Week 2</a><br> | ||
| + | <a href="#July3">July Week 3</a><br> | ||
| + | <a href="#July4">July Week 4</a><br> | ||
| + | <a href="#July5">July Week 5</a><br> | ||
| + | <a href="#Aug1">August Week 1</a><br> | ||
| + | <a href="#Aug2">August Week 2</a><br> | ||
| + | <a href="#Aug3">August Week 3</a><br> | ||
| + | <a href="#Aug4">August Week 4</a><br> | ||
| + | <a href="#Sept1">September Week 1</a><br> | ||
| + | <a href="#Sept2">September Week 2</a><br> | ||
| + | <a href="#Sept3">September Week 3</a><br> | ||
| + | <a href="#Sept4">September Week 4</a><br> | ||
| + | <a href="#Oct1">October Week 1</a><br> | ||
| + | <a href="#Oct2">October Week 2</a><br> | ||
| + | <a href="#Oct3">October Week 3</a><br> | ||
| + | <a href="#Oct4">October Week 4</a><br> | ||
| + | </div> | ||
| + | |||
| + | <p> | ||
| + | |||
| + | |||
| + | <div id="May5"> | ||
<h2>May <br /> | <h2>May <br /> | ||
| - | <br>Week 5<br /></h2> | + | <br>Week 5: Warming up!<br /></h2> |
| + | <h7> | ||
| + | </p> | ||
<ul> | <ul> | ||
| - | <li> | + | <li> We met with our mentors and got an overview of the basics of synthetic biology, namely characterization, which will be the focus of our works during the summer. We also went through the “MoClo” technique, which we intend to introduce to iGEM.</li> |
</ul> | </ul> | ||
| - | |||
| - | |||
<ul> | <ul> | ||
<li>We did a survey of previous iGEM teams that got the Best Experimental Measurements award, then presented the findings to our mentors.</li> | <li>We did a survey of previous iGEM teams that got the Best Experimental Measurements award, then presented the findings to our mentors.</li> | ||
</ul> | </ul> | ||
| + | </h7> | ||
<br> | <br> | ||
| + | |||
| + | |||
| + | <div id="June1"> | ||
<h2>June<br /> | <h2>June<br /> | ||
<br>Week 1<br /></h2> | <br>Week 1<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li>We performed PCR to amplify pMJSAF(LacZ) using the following MoClo forward and reverse primers: DVL0_AF with DVL0_BR (MF1), DVL0_BF with DVL0_CR (MF2), DVL0_CF with DVL0_DR (SJ1) and DVL0_DF with DVL0_ER (SJ2). We performed Gel Electrophoresis to confirm that pMJSAF was amplified in PCR: The gel ran for too long thus the bands ran off of the gel. which can be seen towards the bottom of the picture. We expect for the band to be around 500 bp.</li> | ||
| + | </ul> | ||
| + | <p> | ||
| + | |||
| + | <img src="https://static.igem.org/mediawiki/2012/0/0d/Gel1.png" width="300px"></p> | ||
| + | </ul> | ||
| + | <ul type="disc"> | ||
| + | <li>We reperformed the PCR amplification of pMJSAF with the same pairs of primers: DVL0_AF with DVL0_BR (MF3), DVL0_BF with DVL0_CR (MF4), DVL0_CF with DVL0_DR (SJ3), DVL0_DF with DVL0_ER (SJ4). We performed Gel Electrophoresis on this set of DNA.</li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/1NANV.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>The amplification of pMJSAF displayed strong bands at 500 bp for the primers : DVL0_BF with DVL0_CR and DVL0_DF with DVL0_ER. However for the DVL0_AF with DVL0_BR showed a much weaker band, and the DVL0_CF with DVL0_DR did not show at all. We gel extracted MF3,4 and SJ4. Using the gel extraction protocol. We then quantified the amount of DNA we had using NanoDrop DNA quantification protocol andset up a restriction digest (SpeI, BSA, Buffer 4)for MF3,4 and SJ4. following the Restriction digest protocol </li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/zUbuZ.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>We did PCR clean up of MF3,4 and SJ4 and We quantified the product of MF3,4 and SJ4 PCR clean up using NanoDrop</li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/uli0v.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>For the amplification that didnìt work, we performed gradient PCR of SJ3 and MF3 in order to set the best parameters for the amplification (62-69 with interval of 1 degree C). We later found out, that our step 1 was set at 62C instead of 95C, which could have affected denaturation of the DNA template; although the SJ5-12 was amplified while the MF5-12 did not. We are unsure to the cause. The results were inconclusive compared to the Gel made previously.We ran another PCR to verify results.</li> | ||
| + | <img src="http://i.imgur.com/5IWSS.png" width="300px"></li> | ||
| + | <li>We were given new primers to make stock and working solutions from. In the process we realized that we used DVL1_ER instead of DVL0_ER in SJ4, thus all SJ4 products were discarded and data obsolete. We remade the DVL1_ER stock solution by adding additional EB buffer till the right concentration has been reached. We then proceeded to make stock and working solutions for: DVL0_ER, DVL1_AF, DVL1_ER, DVL1_FF, DVL1_FR, DVL1_GF, DVL1_GR and DVL1_HR. Then we paired AF-ER, FF-GR, EF-FR, GF-HR to amplify the template DNA using PCR. We made the gel from the previously mentioned PCR of AF-ER (MS4), FF-GR (MS5), EF-FR(MS6), GF-HR (MS7) and also 3 L0 (MF3=MS1, SJ3=MS2, SJ4=MS3) <br> | ||
| + | <img src="http://i.imgur.com/woENf.jpg" width="300px"></li> | ||
| + | </ul> | ||
| + | <ul type="disc"> | ||
| + | <li>Alll the L0 failed and the L1 showed bands</li> | ||
| + | <li>We performed gel extraction on the 4 L1 primer based amplification. We performed nanodrop quantification of the products of the gel extraction. </li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/UmCFy.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>We performed restriction digest (SpeI, BSA, Buffer 4)of the products of the gel extraction.From the restriction digest of MS4-7 we performed PCR clean up. We nano dropped the PCR clean up product. </li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/tsl6e.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>We transformed iGEM plasmids with competent bacteriafrom stock, and plated them. We grew them overnight in 37C incubator: pSB1A3 plate 1 well 1G (1A3), pSB1AT3 plate 1 well 13A (1AT3), pSB1C3 plate 1 well 3A (1C3), pSB1K3 plate 1 well 5A (1K3). We picked colonies from the plates that were stored in the incubator and allowed the picked colonies to be incubated in a 37C shaker. We performed miniprep on 3 of the 4 overnight cultures(1k3 was not used due to minimal growth while 1A3, 1AT3 and 1C3 were able to grow sufficiently). We performed nanodrop quantification on the products of the miniprep and then performed restriction digest (SpeI, BSA, Buffer 4)of the products of the miniprep.</li> | ||
| + | </ul> | ||
| + | <ul type="disc"> | ||
| + | <li> We made a second generation of culture of the plasmid carrying bacteria by injecting 20 uL of the original culture into 5 mL of fresh LB broth with the appropriate antibiotics. Using the generation 2 culture of the bacteria with the 1K3 plasmid, we mini prepped it. We nanodrop quantified the mini prepped material. We then ran restriction digest(SpeI, BSA, Buffer 4) of the mini prepped sample.</li> | ||
| + | </ul> | ||
| + | <p><img src="http://i.imgur.com/pPLy8.png" width="300px"></p> | ||
| + | <ul type="disc"> | ||
| + | <li>We streaked plates from the overnight cultures of transformed bacteria (psB1A3, pSB1AT3, pSB1C3, pSB 1k3) and placed them into the 37C incubator.</li> | ||
| + | <li>We prepared a presentation of our project for Wellesley college, whom we will be collaborating with during the summer :)</li> | ||
| + | <li>We had a meeting with professor Doug and other undergrads from CIDAR lab.</li> | ||
| + | </ul> | ||
| + | <br> | ||
| + | <div id="June2"> | ||
| + | <h2>June<br /> | ||
| + | <br>Week 2<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li> | ||
| + | Attended meeting with Wellesley team. Collaboration has started!</p> | ||
| + | </li> | ||
| + | <li> | ||
| + | <p dir="ltr">We performed PCR amplification (20 μL reactions) with new sets of L0 primers: DVL0_EF with DVL0_BR (MS8), DVL0_FF with DVL0_BR (MS9), DVL0_GF with DVL0_BR (MS10), DVL0_DF with DVL0_FR (MS11), DVL0_DF with DVL0_GR (MS12) and DVL0_DF with DVL0_HR (MS13). We then performed gel electrophoresis of MS8-13, all were successful.</p> | ||
| + | </li> | ||
| + | |||
| + | <p><img src="http://i.imgur.com/gUXLG.png" width="300px"></p> | ||
| + | </li> | ||
| + | <li>We performed Gel extraction and nanodropped the samples. </li> | ||
| + | <p><img src="http://i.imgur.com/rx3V2.png" width="300px"></p> </li> | ||
| + | <li>Ran 50 μL PCR reactions of MS8.1-13.1, the same samples as MS8-3, in order to have enough DNA for restriction digest. We performed Gel Electrophoresis of the PCR samples and we performed Gel Extraction. Then, we nanodropped the samples. </li> | ||
| + | <p><img src="http://i.imgur.com/d1aOL.png" width="300px"></p> </li> | ||
| + | <p><img src="http://i.imgur.com/G04xt.png" width="300px"><p> </li> | ||
| + | <li>We performed Restriction digest with the samples MS9.1 and MS 12.1 that had enough DNA concentration. We PCR clean up-ed MS9.1 and 12.1</li> | ||
| + | </ul> | ||
<ul> | <ul> | ||
| - | <li>We performed PCR | + | <li> |
| - | < | + | <p dir="ltr">We performed a new PCR in triplicate for the samples MS8.1, 10.1, 11.1 and 13.1 to achive a higher concentration of DNA. We ran gel of MS8.1, 10.1, 13.1,11.1 to amplify more DNA template to achieve sufficient DNA concentration upon gel extraction</ ></p> |
| - | < | + | </li> |
| - | + | ||
| - | + | <p dir="ltr"> <p><img src="http://i.imgur.com/1A2tt.png" width="300px"</p> | |
| - | + | </li> | |
| - | </ | + | <p><img src="http://i.imgur.com/tXNsB.png" width="300px"</p></li> |
| + | <li>We performed ligation of the successful amplified samples of MS4-7 and MF4: MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3 and MF4 with backbone of 1K3 </li> | ||
</ul> | </ul> | ||
| - | |||
<ul> | <ul> | ||
| - | <li> | + | <li> Ran MgCl2 gradient of MF3 PCR to troubleshoot, along with DMSO separately. <br /> |
| + | </li> | ||
</ul> | </ul> | ||
<ul> | <ul> | ||
| - | + | <li>We transformed the ligations:MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3, MF4 with backbone of 1K3 with competent bacterial cells. We spread plated them with 20 μL of a 40 mg/mL X-Gal solution and 8 μL of a 0.5 IPTG solution for blue-white screening. </li> | |
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <br> | ||
| + | <div id="June3"> | ||
| + | <h2>June<br /> | ||
| + | <br>Week 3<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | <li>We | + | <li> |
| + | We found out that MS8-13 are all wrong due to primer dimer. We redid the PCR of all level 0 primers that failed: L0AB (FJ1), L0EB (FJ2), L0FB (FJ3), L0GB (FJ4), L0DF (FJ5), L0DG (FJ6),L0DH (FJ7), L0DE (FJ8), L0CD (FJ9). Instead of running a gel after the PCR, PCR clean up was performed. The product of the clean up was then nanodropped.</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/uVICV.png" width="300px"</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">Then the PCR clean up samples of FJ1-9 underwent restriction digest. We ran gel of digest, bands came out too weak, but all present.</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/ikiBq.png" width="300px"</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">Redid digest with higher amount of DNA. New gel is much more clear</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/6NYsV.png" width="300px"</p> | ||
| + | </p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">We perfomed gel extraction and nanodropped gel extraction product.</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/bNSaO.png" width="300px"</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | |||
| + | <li> <p dir="ltr"> Performed PCR clean up on RD of 1A3, 1AT3, 1C3, 1K3 transformed at week1 and nanodropped</p> | ||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/0hglX.png" width="300px"</p><br /> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr"> Ligated 1K3 with pSMJSAF from samples of FJ1-9. Transformed the ligations on to plates. The plates did not yield positives for the transformation.</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr"> RD the backbones in two sets, one set with just SpeI other with both SpeI and EcoRI. Gel of the RD showed that there were unexpected bands when cut with both enzymes, something not shown on the registry experience pages for the backbones.</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/FZVtQ.png" width="300px"</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">Plated bacteria transformed with the backbones. Picked colonies from the plates. Miniprepped and nanodropped the cultures with the the backbone</p></li> | ||
| + | |||
| + | <p dir="ltr"> <p><img src="http://i.imgur.com/aAWDI.png" width="300px"</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">Made glycerol stocks of 1AT3, 1A3, 1C3, 1K3 both for -20 and -80 celsius</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> | ||
| + | <p dir="ltr">We prepared plate media of Kan, Amp and CAM</p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <br> | ||
| + | <div id="June4"> | ||
| + | <h2>June<br /> | ||
| + | <br>Week 4<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | |||
| + | <p dir="ltr"> We designed primers and gene synthesis template.</p> | ||
| + | </li> | ||
| + | <li> | ||
| + | <p dir="ltr">We worked on Safety Answers for our Wiki.</p> | ||
| + | </li> | ||
| + | <li> | ||
| + | <p dir="ltr">We attended an Eugene Tutorial given by Ernst, the Pos-Doc responsible for Eugene in CIDAR Lab.</p> | ||
| + | </li> | ||
| + | <li> | ||
| + | <p dir="ltr">We worked on Human Practices project.</p> | ||
| + | </li> | ||
| + | <li> | ||
| + | <p dir="ltr">We met the Wellesley and MIT iGEM teams for a Physics lecture in MIT given by Walter Levine, had lunch together and visited the MIT iGEM team lab :) </p> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <br> | ||
| + | <div id="July1"> | ||
| + | <h2>July<br /> | ||
| + | <br>Week 1 - Happy July 4th!<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li> | ||
| + | <p dir="ltr">We transformed plasmids containing the genes, promoters and terminator to be used as templates for the primers that we had designed. The plasmids come from the iGEM kit plates.</p> | ||
| + | </li> | ||
</ul> | </ul> | ||
<ul> | <ul> | ||
<ul> | <ul> | ||
| - | <li> | + | <li>we used bioline cells but didn’t follow the protocols because the cells were more competent so didn’t need to go through heat shock.</li> |
| - | <li> | + | <li>cells sat on bench over the weekend. Colonies will be picked next week!</li> |
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <li>We also autoclaved Broth, Media and DI water to have enough plates for our next round of transformations.</li> | ||
| + | <li>We used to Clotho to input the primers that we had designed into a database.</li> | ||
| + | <ul> | ||
| + | <li>in doing so, we learned how to use Spreadit Features, Spreadit Oligos, Registry Importer, Collection Viewer and Sequence Viewer.</li> | ||
| + | </ul> | ||
| + | <li>We are still waiting for our primers to continue wet lab work.</li> | ||
</ul> | </ul> | ||
| + | <br> | ||
| + | <div id="July2"> | ||
| + | <h2>July<br /> | ||
| + | <br>Week 2<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | <li>We | + | <ul> |
| - | + | <li>We transformed the following parts from the iGEM plates into bioline cells : E1010, C0051, C0040, B0015, I13458, E0030, E0040, C0012, C0071, R0010, R0063, R0079, I13453, C0079, C0080, R0040, R0051, R0062, R0071, C0062. Cultures from transformation were made and then a miniprep was performed on all of them. We ran a gel to confirm the plasmid presence and performed nanodrop to measure DNA concentration. After that, glycerol stocks were made for all of them.</li> | |
| + | <p dir="ltr"> <p><img src="https://static.igem.org/mediawiki/2012/1/1a/Table7.png" width="300px"</p> | ||
| + | <p dir="ltr"> <p><img src="https://static.igem.org/mediawiki/2012/thumb/1/1c/Table8.PNG/800px-Table8.PNG" width="300px"</p> | ||
</ul> | </ul> | ||
| + | <ul> | ||
| + | <li>We created working and stock solutions for the new primers that arrived to use at PCR. We performed PCR using the transformed parts as templates and the primers we designed in order to add moclo fusion sites to them. The PCR conditions were set according to the table below.</li> | ||
| + | <li><p dir="ltr"> <p><img src="https://static.igem.org/mediawiki/2012/3/34/Pcr_table.PNG" width="300px"</p><br /> | ||
| + | <br /> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We confirmed the PCR reactions by running 5μL agarose gels. For samples bigger than 300bp we ran 1% gel, for samples smaller than that we ran 2% gels. </li> | ||
| + | <li><center> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/9/9a/Gel2.PNG" width="300px">   | ||
| + | <img src="https://static.igem.org/mediawiki/2012/f/f0/GEL3.PNG" width="300px">   | ||
| + | <center> | ||
| + | <li> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/5/50/Gel4.PNG" width="300px">   | ||
| + | <img src="https://static.igem.org/mediawiki/2012/thumb/e/e1/Gel5.PNG/588px-Gel5.PNG" width="300px"> | ||
| + | </ul> | ||
| + | <br> <br /> | ||
| + | <br /> | ||
| + | </li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We performed PCR Clean up in the samples: C0051, C0040, C0062, I13458, R0063AB, R0063EB, R0063FB, R0063GB, R0010AB, E1010, E3030, C0012, C0071, C0080, B0015DE, B0015DF, B0015DG, B0015DH, E0040, C0079, pSB1C3, pSB1k3. Then, we nanodropped them. </li> | ||
| + | <li><img src="https://static.igem.org/mediawiki/2012/thumb/d/d6/Nano2.PNG/485px-Nano2.PNG" width="300px"> </li> | ||
| + | </ul> | ||
| + | <br> | ||
| + | </body> | ||
| + | <div id="July3"> | ||
| + | <h2>July<br /> | ||
| + | <br>Week 3<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | + | <li> | |
| - | <li>We | + | |
| - | <li> | + | We performed restriction digest of the samples with SpeI and NEB buffer 4 for 1 hour. Then, we performed PCR clean up of the RD samples and measured the DNA concentration by nanodrop.<img src="https://lh4.googleusercontent.com/TqhLBBbBB-RQ7-rwzmMNsEAXpDl3PekLs-g9UXCWJENAGi2Jd4_Fe50FHoRz2miBC6CjYyYvHyI8fIS6p-xlwTKVp1OfG5i1L1eajSgs8iwlSgjXCbI" alt="" width="466px;" height="438px;" /><br /> |
| + | </li></ul> | ||
| + | <ul> | ||
| + | <li>We did ligation of pSB1C3 and pSB1k3, in order to circularize the plasmid that we amplified in order to remove RFP.Then, we transformed the ligation product using bioline cells and spread plate them. The 1C3 yielded colonies but the 1K3 didn’t. We picked colonies of 1C3 for overnight culture. <br /> | ||
| + | </li></ul> | ||
| + | <ul> | ||
| + | <li>We performed PCR on pSB1A3, R0010(EB, FB, GB) and R0079 (AB, EB, FB, GB) with phusion from NEB to trouble shoot. For PCR we extended 1A3 extension time, while lowering annealing temperature for all samples. We run a gel to check the PCR product. For 1A3, we ran a 1% gel and for the other samples we ran a 2% gel. Again the amplifications did not work and we decided to double check the primers we designed. We realized that the primers R0010_For_E, R0010_For_F, R0010_For_G, R0079_For_A, R0079_For_E, R0079_For_F an R0079_For_G had problems on their design and couldn`t anneal properly to the template. We are ordering new correct primers and troubleshooting the amplification of pSB1A3.</li> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/9/95/7_19_R0079R0010.PNG" width="300px"> | ||
</ul> | </ul> | ||
| + | <br /> | ||
| + | <ul> | ||
| + | <li>We performed Miniprep in sample pSB1C3 and digested it with SpeI and buffer N4. For the sample pSB1k3 we performed a new ligation, transformed the ligation product using bioline cell and spread plate it. We ran a 1% gel to check the digestion product of pSB1C3 and proceeded to PCR clean up of the sample and DNA quantification.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/f/fc/7_20_1C3.PNG" width="300px"> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/f/ff/Nano.PNG" width="300px"> | ||
| + | <br /> | ||
| + | <br> | ||
| + | <ul> | ||
| + | <li>We started building our level 0 destination vectors :) We ligated our backbone pSB1C3 with the following pcr amplified products: DVL0_GF_BR, DVL0_DF_FR, DVL0_DF_GR, DVL0_CF_DR and DVL0_BF_CR. We transformed them using Bioline competent cells, and plated them with X-gal and IPTG for blue-white screening.</li> | ||
| + | <li>We updated our <a href="https://2012.igem.org/Team:BostonU/Clotho">Clotho experience section</a>, check it out! | ||
| + | |||
| + | </ul> | ||
| + | <br> | ||
| + | </body> | ||
| + | <div id="July4"> | ||
| + | <h2>July<br /> | ||
| + | <br>Week 4<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | + | <li> | |
| - | + | We made cultures of E1010 and pSB1C3, pSB1k3 did not work.</li> | |
| - | + | We transformed ligations of DVL0_GF_BR, DVL0_DF_FR, DVL0_DF_GR, DVL0_CF_DR and DVL0_BF_CR to the pSB1C3 backbone following the bioline cell protocol for ligations because the last set of transformations didn`t work without heat shock. The cells grew but they were not blue. We thought that it might be related to the fact that we did not leave enough time for IPTG and X-Gal be absorbed into the plate. We made a new set of ligations and repeated the transformations, this time placing the plates with IPTG and X-Gal into the incubator for 30 min before plating the cells. And it finally worked! All of them had blue colonies, except for DVL0_FF_BR. </li> | |
| - | + | <li>In order to perform new ligations with the Level 0 inserts, we repeated the protocol from last week to obtain more backbone of pSB1C3.</li> | |
| - | + | <li>Also, we worked on the pSB1k3 backbone. We ran the PCR using the conditions we previously set. We performed PCR clean up AND DNA quantification and ran a gel to confirm the amplification. </li> | |
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/d/d9/2012_7_26_PSB1K3CIRCtrial_.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>We performed a single restriction digest (SpeI) of the sample, DNA quantified it and ligated to itself in order to get circularized backbone.</li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We transformed the ligation product following the Alpha Select cells protocol. The colonies grew! We picked colonies and incubated them overnight. We performed Miniprep and DNA quantification.</li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We ran a single restriction digest (SpeI) and a double restriction digest (SpeI and EcoRI) of the pSB1k3 sample. We ran gel of the double cut and the uncut to confirm the circularized backbone.</li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>Because the transformations of pSB1C3 yielded low count of blue colonies we decided to dephosphorylate the pSB1K3 backbone before ligation in order to decrease the likelihood of the backbone re-ligating to itself. We, then performed PCR clean up and Dna quantification.</li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We, then, ligated the pSB1K3 backbone to the DVL1_ AF_ER, DVL1_ FF_GR, DVL1_ EF_FR and DVL1_ GF_HR inserts we had amplified before. We transformed using the same method as with the Level 0 transformations. | ||
| + | </ul> | ||
| + | <br> | ||
| + | </body> | ||
| + | <div id="July5"> | ||
| + | <h2>July<br /> | ||
| + | <br>Week 5<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | + | ||
| - | + | ||
| - | <li>We | + | <li>We ran a restriction digest with BsaI on miniprep samples from DVL0s transformations of last week to check sizes and send them for sequencing. We ran a ran a gel to confirm it. </li> |
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/thumb/9/92/2012_7_30_RDCUTVUNCUT_.png/442px-2012_7_30_RDCUTVUNCUT_.png" width="300px"><br /> <br /> | ||
| + | <br /> | ||
| + | <ul> | ||
| + | <li>Unfortunately, the sequences we got back showed multiple plasmids contamination. Two of them worked and had a clean sequencing file but lacZ had the wrong orientation. We had to work on the DVL0s again :(</li> | ||
| + | </ul> | ||
| + | <br /> | ||
| + | <ul> | ||
| + | <li>To resequence the DVL0s, we learned in depth on how to streak plates and re streaked plates of the DVL0 cells in order to get single blue colonies. Then, we picked the colonies and ran minipreps on them the other day and made glycerol stocks.</li> | ||
| + | </ul> | ||
| + | <br /><img src="https://static.igem.org/mediawiki/2012/c/c6/76_table.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>We kept working in pSB1K3 backbone, in order to perform new ligations of DVL1. We ran a restriction digest of the psb1k3 backbone with SpeI and ran gel of the RD product with the uncut psB1K3</li> | ||
| + | </ul> | ||
| + | <br /><img src="https://static.igem.org/mediawiki/2012/7/7f/731-12.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>We gel extracted the RD product of pSB1K3, it`s concentration was 5 ng/uL. We, then, dephosphorylated pSB1K3 and ran a PCR clean up with concentration of 5 ng/uL. We finally ligated all the L1s amplified lacZ with psb1K3, doing two transformations for each set, one using dephosphorylated backbone and the other, regular backbone. And they finally grew!!!! :) :)</li> | ||
| + | </ul> | ||
| + | |||
| + | <ul> | ||
| + | <li>We picked blue colonies and let them grow overnight. On the other day, we ran minipreps and made glycerol stocks for them.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/5/53/Table75.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>Besides the struggle with DVs, we proceeded with the PCR of the parts to add MoClo and Restriction sites to them. We ran PCR of J23100 to J23109 primers with circular pSB1C3 template. Ran gel to confirm, but none showed correct bands.</li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li>We ran PCR of J23105-J3108 primers with cut 1C3 backbone hoping that would improve the result and we ran a gel to confirm. Again, no success</li> | ||
| + | </ul> | ||
| + | |||
| + | <ul> | ||
| + | <li>Then, we performed PCR with J23112 primer on uncut 1C3 backbone. We extended the annealing time from 15 seconds to 30 seconds and also decreased the annealing temperature 4 degrees celsius below the indicated temperature on the IDT sheet as an attempt to improve the primers performance. Again, no success. </li> | ||
| + | </ul> | ||
| + | </body> | ||
| + | <div id="Aug1"> | ||
| + | <h2>August<br /> | ||
| + | <br>Week 1<br /></h2> | ||
| + | <h7> | ||
<ul type="disc"> | <ul type="disc"> | ||
| - | <li>We | + | <li>We ran a restriction digest of all the DVL1 and DVL0 miniprep samples (42 in total!!) cut with BsaI, and ran them in gel with their uncut counterparts to check sizes before sending them for sequencing.</li> |
| - | + | <li>We proceeded our troubleshooting PCR of the J and B series using sewing/ligation PCR, a 10 cycle PCR reaction amplifying pSB1C3 and transforming it directly. For more details, check the Methodology section for PCR strategies and troubleshooting. In the beginning, the ligation PCR didn`t work, so we tried a gradient temperature. It showed faint bands but enough to move on and improve it. /li> | |
| - | + | <img src="https://static.igem.org/mediawiki/2012/f/fc/J23105-23108fai.png" width="300px"><br /> | |
| - | + | <li>We realized that non specific bands may be due to excess of DNA template. We ran the PCR again with the decreased amount of template and they worked. Finally :) :) Currently we have J23100-J23112 and J23110-J23118BA-BE, except for J23113 BA. The gel pictures for the amplification can be seen below. </ul> | |
| - | + | <img src="https://static.igem.org/mediawiki/2012/0/0e/J23105-23108suc.png" width="300px"><br /> | |
| - | + | <br> | |
| - | + | </body> | |
| - | + | <div id="Aug2"> | |
| - | + | <h2>August<br> | |
| - | + | <br>Week 2<br /></h2> | |
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li>For the DVs we submitted for sequencing last week, we checked the sequences by blasting them against the expected fragment. All of them worked, except for the DVL0s: CFDR, DFER, FFBR and GFBR that showed the wrong MoClo sites. We had to begin again the process of making these DVL0s :we redid the PCR for them, performed a restriction digest with SpeI, ran a gel, gel extracted them and quantified them.</strong></li> | ||
</ul> | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/3/3f/M91nano.PNG" width="300px"><br/> | ||
| + | <ul> | ||
| + | <li> We ligated those parts to 1C3 backbone and transformed them. Unfortunately, the cells we used the first time were not so competent anymore and the transformations didn't work. So, we repeated the transformations with Bioline cells and let them growing on the bench over the weekend.</strong></strong> </li> | ||
</ul> | </ul> | ||
| - | <ul | + | <ul> |
| - | <li> | + | <li> After using pSB1C3 for the ligations, we were running short of the backbone to use as template for ligation PCR. So we streaked plates from glycerol stocks, pick colonies and ran miniprep on the other day. Then, we ran a RD, pcr clean up and quantified it. </strong></li> |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/c/cb/M95nano.PNG" width="300px"><br /<br /> | ||
| + | </strong> | ||
<ul> | <ul> | ||
| - | + | <li> We did our first set of MoClo reactions in order to make L0 MoClo parts.</strong></li> | |
| - | <li> | + | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | |||
| + | <ul> | ||
| + | <li> We transformed them and let them growing on the bench over the weekend to perform blue-white screening next week. </strong></li> | ||
</ul> | </ul> | ||
| - | <br> | + | <br /> |
| - | < | + | </strong> |
| - | + | ||
<ul> | <ul> | ||
| - | <li> | + | <li> Also, we started working on our DVL2s. We are using amp as our resistance backbone, so we decided to get it from one of miniprep samples we had from C0040 that has an amp backbone. We ran RD of C0040 with XbaI and SpeI to get C0040 out, we ran a gel to confirm it and gel extracted it.</strong></li> |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/c/c2/2012_8_15_psb1a2C0040.png" width="300px"><br /> | ||
| + | </strong> | ||
| + | <ul> | ||
| + | <li> Then, we performed PCR using pSB1A2 as template and BBpls_For and BBpls_Rev as primers. We ran a gel to confirm it and gel extracted it.</strong></li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/6/6b/2012_8_16_psb1a2%2Bpcr3%281%29.png" width="300px"><br /> | ||
| + | </strong> | ||
| + | <ul> | ||
| + | <li> Then, we ran Restriction digest with SpeI, PCR clean up and its concentration was 3.2 ng/uL. DVL2 backbone is ready to be ligated with amplified LacZ having the MoClo sites :)</ul> | ||
| + | <br> | ||
| + | </body> | ||
| + | <div id="aug3"> | ||
| + | <h2>August<br> | ||
| + | <br>Week 3<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li>We kept working on L2 DVs. We performed a ligation of backbone pSB1A2 to itself and transformed the circularized backbone. We picked colonies the following and set overnight cultures of the plasmid. Then, we performed minipreps and got 28 ng/uL DNA concentration. Also, we amplified LacZ with the following primers: DVL0_For_A and DVL0_Rev_F, DVL0_For_A and DVL0_Rev_G, DVL0_For_A and DVL0_Rev_H, DVL0_For_E and DVL0_Rev_G, DVL0_For_E and DVL0_Rev, H and DVL0_For_F and DVL0_Rev_H. We digested both the LacZ amplified fragments and the pSB1A2 backbone with SpeI cut. We ran the cut sample son a gel to confirm sizes.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/4/42/2012_8_23_RD_111_.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We performed gel extraction and then, quantified the samples. </li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/thumb/e/e2/122_tsable.png/800px-122_tsable.png" width="300px"><br /><br /><br /> | ||
| + | <ul> | ||
| + | <li>We performed ligation of the DVL2_AF_BR, DVL2_AF_GR, DVL2_AF_HR, DVL2_EF_GR and DVL2_EF_HR with pSB1A2 and transformed them with bioline gold cells for blue-white screening. Unfortunately, colonies grew but none of them turned blue. </li> | ||
| + | <li>We dephosphorylated the backbone pSB1A2 to prevent recircularization when ligating it to amplified LacZ. We performed new ligation and transformed them. Again, colonies grew but none of them turned blue. We are troubleshooting the experiment now. </li> | ||
| + | <li>The MoClo L0 parts transformations worked nicely, plates showed more white colonies then blue, indicating that LacZ was cut out of the backbone and the Moclo parts took place. We picked white colonies and let them grow overnight. We performed miniprep in all of them and Dna quantified them.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/thumb/5/55/122_tsaddddble.png/297px-122_tsaddddble.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>In order to check the MoClo parts before send it for sequencing, we ran BsaI digests and ran the digest product on a gel. </li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/8/84/122_tsaddble.png"width=" 300px"> | ||
| + | |||
| + | <div id="Aug4"> | ||
| + | <h2>August<br /> | ||
| + | <br>Week 4<br /></h2> | ||
| + | <h7> | ||
| + | <ul type="disc"> | ||
| + | <li>We continued the ligation PCRs to add MoClo sites to J series promoters, RBSs and genes. We tried to scale down the first set of reactions, because we were only using 2 uL of the reaction product as template for third set of pcr reactions. It didn`t work, however. The gel showed really faint bands and it was impossible to gel extract them.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/4/4d/2012_8_25_pcr_117.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We performed new 50 ul PCRs for J23103-J23106 BA-BE.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/0/05/2012_8_28_pcrpromoter_series_119.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>Also, we ran regular PCRs to amplify the genes with correct primers that arrived recently: R0010EB, R0010FB, R0010GB, R0079AB, R0079EB, R0079FB, R0079GB and I1453FB. </li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/6/69/2012_8_25_pcrpromoter_series_118.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We, then, gel extracted the bands and quantified the DNA from both gels.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/2/24/120table.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>Also, we performed ligation PCR for B0030-34 BC.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/c/c2/2012_8_29_pcrrbsseries_123.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>Then, we gel extracted and quantified it.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/thumb/4/4e/Table124.png/800px-Table124.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>After using so much pSB1C3 as template for PCR we ran out of it and had to streak plates from the Glycerol stocks, run minipreps, digest, clean up and finally continue with our PCRs :) We ran ligation PCR of J23107-09 AB-EB, J23119AB-EB and J23113AB, J23103AB.</li> | ||
| + | </ul> | ||
| + | |||
| + | <body> | ||
| + | <div id="Sept1"> | ||
| + | <h2>September <br /> | ||
| + | <br>Week 1<br /></h2> | ||
| + | <ul> | ||
| + | <li>We ran a gel to check the ligation PCRs from last week: 23107-09 AB-EB, J23119AB-EB and J23113AB. Then, we performed gel extraction.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/c/ca/09_28_12_top_level1p128.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>We quantified the samples</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/9/91/P86sA.png" width="300px">.<br /> | ||
| + | <ul> | ||
| + | <li>We also repeated the PCR of lacZ with to add the fusion sites and build our DVL2s. We ran a PCR clean up and quantified the samples.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/a/a2/P86sU111.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We digested the samples with SpeI and ran a gel of the digested samples to check sizes. Then, we ran a gel extraction.</li> | ||
| + | </ul> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/8/86/2012_9_10_dvl2p129_.png" width="300px"> | ||
| + | <ul> | ||
| + | <li>And quantified the samples</li> | ||
| + | </ul> | ||
| + | <br /><img src="https://static.igem.org/mediawiki/2012/6/6e/Table_p131.PNG" width="300px"> | ||
| + | |||
| + | <ul> | ||
| + | <div id="Sept2"> | ||
| + | <h2>September | ||
| + | Week 2</h2> | ||
| + | <ul> | ||
| + | <li>We continued to work with DVL2s. We ran ligation of lacZ amplified with level 2 fusion sites and pSB1A2 as backbone and transformed them. However, they did not grow blue, we could only see white colonies. We tried once more but they are still not growing. We are working on troubleshooting them.</li> | ||
| + | <li>We kept troubleshooting the DVL0s that didn`t work. We picked colonies of DVL0_BFCR plate and miniprepped 3 samples to check the correct sequence. We submmited them for sequencing along with previous MoClo reactions 1 and 14-22. We continued with the MoClo reactions and we ran reactions 23-49.</li> | ||
| + | </ul> | ||
| + | <br /><img src="https://static.igem.org/mediawiki/2012/4/4f/23-49.PNG" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We transformed all MoClo reactions (23-49) with Zymo cells. Unfortunately, the cells were not very competent and none of plates grew sufficiently. We tried transforming once more with Bioline Gold Cells, this time we included Moclo 50-54 .</li> | ||
</ul> | </ul> | ||
| + | table moclo 50-54.<br /><img src="https://static.igem.org/mediawiki/2012/8/85/50-541ss1.PNG" width="100px"> | ||
<ul> | <ul> | ||
| + | <li>We started our first Flow Cytometry experiments. In the first day, we streaked plates for getting fresh colonies. The samples were:</li> | ||
<ul> | <ul> | ||
| - | <li> | + | <li>Amp: GFP+, RFP+, lacI->pLacI(IPTG curv), TetR->pTetR(aTe Curve), TetR inverter (Ara curve), LuxR->pLuxR(AHL curve)</li> |
| - | + | <li>Negative control</li> | |
| - | <li> | + | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| + | <li>In the second day, we picked colonies for flow cytometry in triplicates and let them growing in 2 mL of culture for 6 hours. After that, we added: 199ul of LB with proper antibiotic and inducer/ repressor molecules to tubes and innoculated 1 uL of the culture and let them growing overnight. </li> | ||
| + | <li>In the third day, we added 450ul of PBS and 50 uL of overnight cultures to the Flow Cytometry tubes and ran the experiments. The graphs and results can be seen in Results page. </li> | ||
</ul> | </ul> | ||
<br> | <br> | ||
| - | <h2>Week 3 | + | <div id="Sept3"> |
| + | <h2>September | ||
| + | Week 3</h2> | ||
<ul> | <ul> | ||
| - | + | li>We continued troubleshooting the DVL0s that didn`t work and picked colonies from a DVL0_AF_BR plate and send them for sequencing. We also sent MoClo 2, 4, 5, 7, 8, 9, 10, 11, 12, 13 and 21 for sequencing.</li> | |
| - | + | <li>We transformed another set of Moclo reactions using Bioline Gold cells: 23-29 and 50-54, picked colonies and ran miniprep on them. We picked two colonies for each gene and four colonies for each RBS. We quantified all the 34 minipreps.</li> | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| - | < | + | <img src="https://static.igem.org/mediawiki/2012/e/e9/Table138.png" width="300px"> |
| - | + | <li>We ran more Flow Cytometry experiments. In the first day, we streaked plates for getting fresh colonies. The samples were:pConst + rbs-lacI-GFPc-pLacI-RFPc , pConst + rbs-tetR-GFPc-pTet-RFPc, I13458-I13453+B0032+C0040+B0034+E0040+B0015+R0040+B0034+E1010+B0015, R0040-B0034-C0062-B0015-R0065-J23101, GFP positive control, RFP positive control and negative control. We followed the same protocol from last week and ran the Flow Cytometry experiment.</li> | |
| - | + | <ul> | |
| - | + | <li>We ran more MoClo reactions (55-66) and transformed them using Zymo cells. </li> | |
| - | <li> | + | |
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
</ul> | </ul> | ||
| - | < | + | <img src="https://static.igem.org/mediawiki/2012/f/fb/Moclo55-64.PNG" width="100px"> <br /> |
| - | <li> | + | <ul> |
| + | <li>We picked colonies from MoClo plates (30-39) ran minipreps in the samples and quantified them.</li> | ||
</ul> | </ul> | ||
| - | < | + | <img src="https://static.igem.org/mediawiki/2012/3/38/Kkkkkkkkkkk.png" width="300px"> </strong> |
| - | </ | + | </body> |
| + | |||
| + | </ul> | ||
| + | <br> | ||
| + | <div id="Sept4"> | ||
| + | <h2>September | ||
| + | Week 4</h2> | ||
<ul> | <ul> | ||
| - | + | <h7><li>We sent 53 MoClo reactions for sequencing: MoClo23- MoClo 54, with duplicates for genes and quadruples for RBSs.</strong></li> | |
| - | <li> | + | |
| - | <li> | + | </ul> |
| - | <li> | + | <ul> |
| - | <li> | + | <li>We transformed them using Zymo cells and overloaded the reaction using 50 uL of cells and 4 uL of DNA. We picked 4 colonies for each transformation the next day and ran a small diges with X,P to check the sizes. However, they were incorrect, and we are in the process of troubleshooting.</strong></li> |
| - | <li> | + | </ul> |
| - | <li> | + | <ul> |
| - | <li> | + | <li>We quantified the ones that were correct and streaked plates for running Flow Cytometry experiments. </strong></li> |
| - | <li> | + | </ul> |
| - | <li> | + | <br /> |
| - | <li> | + | <ul> |
| - | </li> | + | <li>For the Level 0 MoClo parts that didn`t show the correct sequences we picked two new colonies from each plate we had. MoClo 36, 37, 38, 39, 40, 10, 32, 27, 26, 6, 7, 8, 2, 3, 29, 23, 24, 30, 11, 43, 44, 16, 45, 4, 47, 33, 48 and 49. We ran small digest with x and p and ran them on a gel to check the sizes.</strong></li> |
| - | <li>We | + | </ul> |
| - | <li> | + | <img src="https://static.igem.org/mediawiki/2012/d/dc/144_top.png" width="300px"> |
| + | <img src="https://static.igem.org/mediawiki/2012/c/c8/144mid.png" width="300px"> | ||
| + | <img src="https://static.igem.org/mediawiki/2012/2/29/144bot.png" width="300px"><br /> | ||
| + | <ul> | ||
| + | <li>We submitted the samples that seemed to show the correct band for sequencing. </strong></li> | ||
| + | </ul> | ||
| + | <ul> | ||
| + | <li> Also, we worked on streaking plates for the samples we are submitting to the registry.</strong></li> | ||
| + | </ul> | ||
| + | |||
| + | <div id="Oct1"> | ||
| + | <h2>October | ||
| + | Week 1</h2> | ||
| + | <ul> | ||
| + | <li>We miniprepped the samples we are sending to the registry and they are ready for submission :) The list of parts can be seen in the section Parts Submitted. </li> | ||
| + | <li>We picked colonies for the Flow Cytometry Experiment: All the Moclo L1 parts that worked (L1.2, L1.4, L1.5, L1.6, L1.7, L1.8 L1.10) along with their Biobrick counterparts. We ran the Flow Cytometry Experiments and the results can be checked at the section Results Summary. </li> | ||
| + | <li>We went over MIT to drop off the samples for the Registry.</li> | ||
| + | <li>Wiki is freezed! See you at the Jamboree :)</li> | ||
| + | </ul> | ||
| + | |||
| + | <div id="Oct2"> | ||
| + | <h2>October <br /> | ||
| + | <br>Week 2<br /></h2> | ||
| + | |||
| + | <ul> | ||
| + | <h7> <li>One problem that we found out was keeping us from creating Level1 parts was because we mislabeled tubes, and were using incorrect Level 1 destination vectors to ligate with the parts. </li> | ||
| + | <li>We transformed with bioline cells level 2 destination vectors, but there were not many blue colonies upon blue white screening.</li> | ||
| + | <li>We continued to convert biobrick promoters of the J series into level 0 moclo parts and also set up moclo level 1 reactions. They were transformed and picked using blue white screening. The next day, we prepared cultures and then ran prep. We checked the constructs by running digest with X,P for MoClo level 0 and level 1, then we ran gel to confirm.</li> | ||
| + | <li><p><img src="https://static.igem.org/mediawiki/2012/3/37/2012_10_8_Level0_Digest_bottom_.png" width="300px"></p></li> | ||
| + | <li><p><img src="https://static.igem.org/mediawiki/2012/e/e2/2012_10_8_Level0_Digest_top_.png" width="300px"></p></li> | ||
| + | <li>The correct bands were then sequenced and they yielded the correct orientation and nucleotide order in the forward direction.</li> | ||
| + | <li>In terms of troubleshooting DVL2s, we decided to remake the pSB1A2 backbone from a glycerol stock of the biobrick of C0080 and digested it with X,P, in order to cut out the part. </li> | ||
| + | <li>And we are going to the iGEM Regionals at Pittsburgh! </li> | ||
| + | </ul> | ||
| + | <div id="Oct3"> | ||
| + | <h2>October <br /> | ||
| + | <br>Week 3<br /></h2><ul> | ||
| + | |||
| + | <li>We are very excited to be back in the lab and work for the Worlds Jamboree!! :)</li> | ||
| + | <li>First, we are taking care of transferring the parts we submitted from pSB1A2 to pSB1C3. We did this by digesting all the parts with E, S and pSB1C3 also. We ligated the digested parts and pSB1C3 and transformed. However, none of them grew. We believe this is due to the low yield of Gel Extraction.</li> | ||
| + | <li>We also added fusion sites to R0040 using ligation PCR, we verified them on the gel and gel extracted it. </li> | ||
| + | |||
| + | <div id="Oct4"> | ||
| + | <h2>October <br /> | ||
| + | <br>Week 4<br /></h2> | ||
| + | |||
| + | <ul><h7><li>We transformed a new set of Moclo Level 0 reactions , We got white colonies for the blue-white screening and we made cultures and ran a prep. However, when we digested them to confirm the sizes none of the parts showed the bands we were expecting. We think this might be due to the amount of colonies we screened for each part, usually we have to screen four colonies per part and we just screened two.</li> | ||
| + | <li>Regarding the MoClo Level 1, we set up new reactions using the Promega buffer. Also, we doubled the volume of BsaI after we realized it was two times lower than predicted. We transformed them but we didn`t get any colonies. We tried new reaction with a higher volume of Ligase and we are waiting for transformation to check them. Fingers crossed :)</li> | ||
| + | <li>Because the initial effort to convert the biobrick parts into the pSB1C3 backbone didn’t work, we tried the entire process again. However, as of Friday 10/26, we haven`t get to the point of transforming the ligations. </li> | ||
| + | <li>In the process of creating level 2 destination vectors, we already isolated the backbone from a biobrick part, we then digested with SpeI and gel extracted it in preparation for ligation with lacZ. </li> | ||
| + | <li><p><img src="https://static.igem.org/mediawiki/2012/2/2e/2012_10_dasdfsdafxa2.jpg" width="300px"></p></li> | ||
| + | |||
| + | <li>Afterwards we ligated Lacz with different sites | ||
| + | <li> New results will included as part of our presentation. See you all at MIT!<br /> | ||
| + | </li> | ||
</ul> | </ul> | ||
| - | |||
</body> | </body> | ||
Latest revision as of 17:47, 26 October 2012

Weekly Notebook
May
Week 5: Warming up!
- We met with our mentors and got an overview of the basics of synthetic biology, namely characterization, which will be the focus of our works during the summer. We also went through the “MoClo” technique, which we intend to introduce to iGEM.
- We did a survey of previous iGEM teams that got the Best Experimental Measurements award, then presented the findings to our mentors.
June
Week 1
- We performed PCR to amplify pMJSAF(LacZ) using the following MoClo forward and reverse primers: DVL0_AF with DVL0_BR (MF1), DVL0_BF with DVL0_CR (MF2), DVL0_CF with DVL0_DR (SJ1) and DVL0_DF with DVL0_ER (SJ2). We performed Gel Electrophoresis to confirm that pMJSAF was amplified in PCR: The gel ran for too long thus the bands ran off of the gel. which can be seen towards the bottom of the picture. We expect for the band to be around 500 bp.

- We reperformed the PCR amplification of pMJSAF with the same pairs of primers: DVL0_AF with DVL0_BR (MF3), DVL0_BF with DVL0_CR (MF4), DVL0_CF with DVL0_DR (SJ3), DVL0_DF with DVL0_ER (SJ4). We performed Gel Electrophoresis on this set of DNA.

- The amplification of pMJSAF displayed strong bands at 500 bp for the primers : DVL0_BF with DVL0_CR and DVL0_DF with DVL0_ER. However for the DVL0_AF with DVL0_BR showed a much weaker band, and the DVL0_CF with DVL0_DR did not show at all. We gel extracted MF3,4 and SJ4. Using the gel extraction protocol. We then quantified the amount of DNA we had using NanoDrop DNA quantification protocol andset up a restriction digest (SpeI, BSA, Buffer 4)for MF3,4 and SJ4. following the Restriction digest protocol

- We did PCR clean up of MF3,4 and SJ4 and We quantified the product of MF3,4 and SJ4 PCR clean up using NanoDrop

- For the amplification that didnìt work, we performed gradient PCR of SJ3 and MF3 in order to set the best parameters for the amplification (62-69 with interval of 1 degree C). We later found out, that our step 1 was set at 62C instead of 95C, which could have affected denaturation of the DNA template; although the SJ5-12 was amplified while the MF5-12 did not. We are unsure to the cause. The results were inconclusive compared to the Gel made previously.We ran another PCR to verify results.
- We were given new primers to make stock and working solutions from. In the process we realized that we used DVL1_ER instead of DVL0_ER in SJ4, thus all SJ4 products were discarded and data obsolete. We remade the DVL1_ER stock solution by adding additional EB buffer till the right concentration has been reached. We then proceeded to make stock and working solutions for: DVL0_ER, DVL1_AF, DVL1_ER, DVL1_FF, DVL1_FR, DVL1_GF, DVL1_GR and DVL1_HR. Then we paired AF-ER, FF-GR, EF-FR, GF-HR to amplify the template DNA using PCR. We made the gel from the previously mentioned PCR of AF-ER (MS4), FF-GR (MS5), EF-FR(MS6), GF-HR (MS7) and also 3 L0 (MF3=MS1, SJ3=MS2, SJ4=MS3)


- Alll the L0 failed and the L1 showed bands
- We performed gel extraction on the 4 L1 primer based amplification. We performed nanodrop quantification of the products of the gel extraction.

- We performed restriction digest (SpeI, BSA, Buffer 4)of the products of the gel extraction.From the restriction digest of MS4-7 we performed PCR clean up. We nano dropped the PCR clean up product.

- We transformed iGEM plasmids with competent bacteriafrom stock, and plated them. We grew them overnight in 37C incubator: pSB1A3 plate 1 well 1G (1A3), pSB1AT3 plate 1 well 13A (1AT3), pSB1C3 plate 1 well 3A (1C3), pSB1K3 plate 1 well 5A (1K3). We picked colonies from the plates that were stored in the incubator and allowed the picked colonies to be incubated in a 37C shaker. We performed miniprep on 3 of the 4 overnight cultures(1k3 was not used due to minimal growth while 1A3, 1AT3 and 1C3 were able to grow sufficiently). We performed nanodrop quantification on the products of the miniprep and then performed restriction digest (SpeI, BSA, Buffer 4)of the products of the miniprep.
- We made a second generation of culture of the plasmid carrying bacteria by injecting 20 uL of the original culture into 5 mL of fresh LB broth with the appropriate antibiotics. Using the generation 2 culture of the bacteria with the 1K3 plasmid, we mini prepped it. We nanodrop quantified the mini prepped material. We then ran restriction digest(SpeI, BSA, Buffer 4) of the mini prepped sample.

- We streaked plates from the overnight cultures of transformed bacteria (psB1A3, pSB1AT3, pSB1C3, pSB 1k3) and placed them into the 37C incubator.
- We prepared a presentation of our project for Wellesley college, whom we will be collaborating with during the summer :)
- We had a meeting with professor Doug and other undergrads from CIDAR lab.
June
Week 2
- Attended meeting with Wellesley team. Collaboration has started!
-
We performed PCR amplification (20 μL reactions) with new sets of L0 primers: DVL0_EF with DVL0_BR (MS8), DVL0_FF with DVL0_BR (MS9), DVL0_GF with DVL0_BR (MS10), DVL0_DF with DVL0_FR (MS11), DVL0_DF with DVL0_GR (MS12) and DVL0_DF with DVL0_HR (MS13). We then performed gel electrophoresis of MS8-13, all were successful.
- We performed Gel extraction and nanodropped the samples.
- Ran 50 μL PCR reactions of MS8.1-13.1, the same samples as MS8-3, in order to have enough DNA for restriction digest. We performed Gel Electrophoresis of the PCR samples and we performed Gel Extraction. Then, we nanodropped the samples.
- We performed Restriction digest with the samples MS9.1 and MS 12.1 that had enough DNA concentration. We PCR clean up-ed MS9.1 and 12.1




-
We performed a new PCR in triplicate for the samples MS8.1, 10.1, 11.1 and 13.1 to achive a higher concentration of DNA. We ran gel of MS8.1, 10.1, 13.1,11.1 to amplify more DNA template to achieve sufficient DNA concentration upon gel extraction
- We performed ligation of the successful amplified samples of MS4-7 and MF4: MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3 and MF4 with backbone of 1K3


- Ran MgCl2 gradient of MF3 PCR to troubleshoot, along with DMSO separately.
- We transformed the ligations:MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3, MF4 with backbone of 1K3 with competent bacterial cells. We spread plated them with 20 μL of a 40 mg/mL X-Gal solution and 8 μL of a 0.5 IPTG solution for blue-white screening.
June
Week 3
- We found out that MS8-13 are all wrong due to primer dimer. We redid the PCR of all level 0 primers that failed: L0AB (FJ1), L0EB (FJ2), L0FB (FJ3), L0GB (FJ4), L0DF (FJ5), L0DG (FJ6),L0DH (FJ7), L0DE (FJ8), L0CD (FJ9). Instead of running a gel after the PCR, PCR clean up was performed. The product of the clean up was then nanodropped.

-
Then the PCR clean up samples of FJ1-9 underwent restriction digest. We ran gel of digest, bands came out too weak, but all present.

-
Redid digest with higher amount of DNA. New gel is much more clear

-
We perfomed gel extraction and nanodropped gel extraction product.

-
Performed PCR clean up on RD of 1A3, 1AT3, 1C3, 1K3 transformed at week1 and nanodropped

-
Ligated 1K3 with pSMJSAF from samples of FJ1-9. Transformed the ligations on to plates. The plates did not yield positives for the transformation.
-
RD the backbones in two sets, one set with just SpeI other with both SpeI and EcoRI. Gel of the RD showed that there were unexpected bands when cut with both enzymes, something not shown on the registry experience pages for the backbones.

-
Plated bacteria transformed with the backbones. Picked colonies from the plates. Miniprepped and nanodropped the cultures with the the backbone

-
Made glycerol stocks of 1AT3, 1A3, 1C3, 1K3 both for -20 and -80 celsius
-
We prepared plate media of Kan, Amp and CAM
June
Week 4
-
We worked on Safety Answers for our Wiki.
-
We attended an Eugene Tutorial given by Ernst, the Pos-Doc responsible for Eugene in CIDAR Lab.
-
We worked on Human Practices project.
-
We met the Wellesley and MIT iGEM teams for a Physics lecture in MIT given by Walter Levine, had lunch together and visited the MIT iGEM team lab :)
We designed primers and gene synthesis template.
July
Week 1 - Happy July 4th!
-
We transformed plasmids containing the genes, promoters and terminator to be used as templates for the primers that we had designed. The plasmids come from the iGEM kit plates.
- we used bioline cells but didn’t follow the protocols because the cells were more competent so didn’t need to go through heat shock.
- cells sat on bench over the weekend. Colonies will be picked next week!
- We also autoclaved Broth, Media and DI water to have enough plates for our next round of transformations.
- We used to Clotho to input the primers that we had designed into a database.
- in doing so, we learned how to use Spreadit Features, Spreadit Oligos, Registry Importer, Collection Viewer and Sequence Viewer.
- We are still waiting for our primers to continue wet lab work.
July
Week 2
- We transformed the following parts from the iGEM plates into bioline cells : E1010, C0051, C0040, B0015, I13458, E0030, E0040, C0012, C0071, R0010, R0063, R0079, I13453, C0079, C0080, R0040, R0051, R0062, R0071, C0062. Cultures from transformation were made and then a miniprep was performed on all of them. We ran a gel to confirm the plasmid presence and performed nanodrop to measure DNA concentration. After that, glycerol stocks were made for all of them.
- We created working and stock solutions for the new primers that arrived to use at PCR. We performed PCR using the transformed parts as templates and the primers we designed in order to add moclo fusion sites to them. The PCR conditions were set according to the table below.

- We confirmed the PCR reactions by running 5μL agarose gels. For samples bigger than 300bp we ran 1% gel, for samples smaller than that we ran 2% gels.






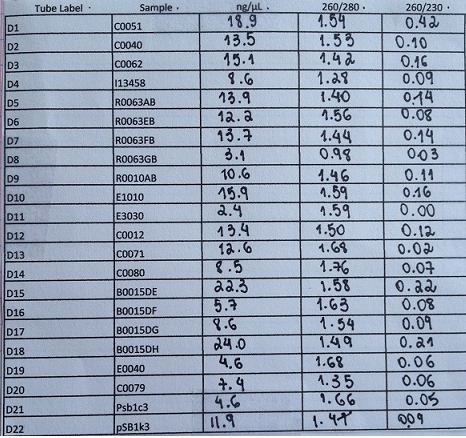
- We performed PCR Clean up in the samples: C0051, C0040, C0062, I13458, R0063AB, R0063EB, R0063FB, R0063GB, R0010AB, E1010, E3030, C0012, C0071, C0080, B0015DE, B0015DF, B0015DG, B0015DH, E0040, C0079, pSB1C3, pSB1k3. Then, we nanodropped them.

July
Week 3
-
We performed restriction digest of the samples with SpeI and NEB buffer 4 for 1 hour. Then, we performed PCR clean up of the RD samples and measured the DNA concentration by nanodrop.
- We did ligation of pSB1C3 and pSB1k3, in order to circularize the plasmid that we amplified in order to remove RFP.Then, we transformed the ligation product using bioline cells and spread plate them. The 1C3 yielded colonies but the 1K3 didn’t. We picked colonies of 1C3 for overnight culture.
- We performed PCR on pSB1A3, R0010(EB, FB, GB) and R0079 (AB, EB, FB, GB) with phusion from NEB to trouble shoot. For PCR we extended 1A3 extension time, while lowering annealing temperature for all samples. We run a gel to check the PCR product. For 1A3, we ran a 1% gel and for the other samples we ran a 2% gel. Again the amplifications did not work and we decided to double check the primers we designed. We realized that the primers R0010_For_E, R0010_For_F, R0010_For_G, R0079_For_A, R0079_For_E, R0079_For_F an R0079_For_G had problems on their design and couldn`t anneal properly to the template. We are ordering new correct primers and troubleshooting the amplification of pSB1A3.

- We performed Miniprep in sample pSB1C3 and digested it with SpeI and buffer N4. For the sample pSB1k3 we performed a new ligation, transformed the ligation product using bioline cell and spread plate it. We ran a 1% gel to check the digestion product of pSB1C3 and proceeded to PCR clean up of the sample and DNA quantification.


- We started building our level 0 destination vectors :) We ligated our backbone pSB1C3 with the following pcr amplified products: DVL0_GF_BR, DVL0_DF_FR, DVL0_DF_GR, DVL0_CF_DR and DVL0_BF_CR. We transformed them using Bioline competent cells, and plated them with X-gal and IPTG for blue-white screening.
- We updated our Clotho experience section, check it out!






















 .
.












