 "
"
Team:BostonU/Notebook
From 2012.igem.org

Weekly Notebook
May
Week 5: Warming up!
- We met with our mentors and got an overview of the basics of synthetic biology, namely characterization, which will be the focus of our works during the summer. We also went through the “MoClo” technique, which we intend to introduce to iGEM.
- We did a survey of previous iGEM teams that got the Best Experimental Measurements award, then presented the findings to our mentors.
June
Week 1
- We performed PCR to amplify pMJSAF(LacZ) using the following MoClo forward and reverse primers: DVL0_AF with DVL0_BR (MF1), DVL0_BF with DVL0_CR (MF2), DVL0_CF with DVL0_DR (SJ1) and DVL0_DF with DVL0_ER (SJ2). We performed Gel Electrophoresis to confirm that pMJSAF was amplified in PCR: The gel ran for too long thus the bands ran off of the gel. which can be seen towards the bottom of the picture. We expect for the band to be around 500 bp.

- We reperformed the PCR amplification of pMJSAF with the same pairs of primers: DVL0_AF with DVL0_BR (MF3), DVL0_BF with DVL0_CR (MF4), DVL0_CF with DVL0_DR (SJ3), DVL0_DF with DVL0_ER (SJ4). We performed Gel Electrophoresis on this set of DNA.

- The amplification of pMJSAF displayed strong bands at 500 bp for the primers : DVL0_BF with DVL0_CR and DVL0_DF with DVL0_ER. However for the DVL0_AF with DVL0_BR showed a much weaker band, and the DVL0_CF with DVL0_DR did not show at all. We gel extracted MF3,4 and SJ4. Using the gel extraction protocol. We then quantified the amount of DNA we had using NanoDrop DNA quantification protocol andset up a restriction digest (SpeI, BSA, Buffer 4)for MF3,4 and SJ4. following the Restriction digest protocol

- We did PCR clean up of MF3,4 and SJ4 and We quantified the product of MF3,4 and SJ4 PCR clean up using NanoDrop

- For the amplification that didnìt work, we performed gradient PCR of SJ3 and MF3 in order to set the best parameters for the amplification (62-69 with interval of 1 degree C). We later found out, that our step 1 was set at 62C instead of 95C, which could have affected denaturation of the DNA template; although the SJ5-12 was amplified while the MF5-12 did not. We are unsure to the cause. The results were inconclusive compared to the Gel made previously.We ran another PCR to verify results.
- We were given new primers to make stock and working solutions from. In the process we realized that we used DVL1_ER instead of DVL0_ER in SJ4, thus all SJ4 products were discarded and data obsolete. We remade the DVL1_ER stock solution by adding additional EB buffer till the right concentration has been reached. We then proceeded to make stock and working solutions for: DVL0_ER, DVL1_AF, DVL1_ER, DVL1_FF, DVL1_FR, DVL1_GF, DVL1_GR and DVL1_HR. Then we paired AF-ER, FF-GR, EF-FR, GF-HR to amplify the template DNA using PCR. We made the gel from the previously mentioned PCR of AF-ER (MS4), FF-GR (MS5), EF-FR(MS6), GF-HR (MS7) and also 3 L0 (MF3=MS1, SJ3=MS2, SJ4=MS3)


- Alll the L0 failed and the L1 showed bands
- We performed gel extraction on the 4 L1 primer based amplification. We performed nanodrop quantification of the products of the gel extraction.

- We performed restriction digest (SpeI, BSA, Buffer 4)of the products of the gel extraction.From the restriction digest of MS4-7 we performed PCR clean up. We nano dropped the PCR clean up product.

- We transformed iGEM plasmids with competent bacteriafrom stock, and plated them. We grew them overnight in 37C incubator: pSB1A3 plate 1 well 1G (1A3), pSB1AT3 plate 1 well 13A (1AT3), pSB1C3 plate 1 well 3A (1C3), pSB1K3 plate 1 well 5A (1K3). We picked colonies from the plates that were stored in the incubator and allowed the picked colonies to be incubated in a 37C shaker. We performed miniprep on 3 of the 4 overnight cultures(1k3 was not used due to minimal growth while 1A3, 1AT3 and 1C3 were able to grow sufficiently). We performed nanodrop quantification on the products of the miniprep and then performed restriction digest (SpeI, BSA, Buffer 4)of the products of the miniprep.
- We made a second generation of culture of the plasmid carrying bacteria by injecting 20 uL of the original culture into 5 mL of fresh LB broth with the appropriate antibiotics. Using the generation 2 culture of the bacteria with the 1K3 plasmid, we mini prepped it. We nanodrop quantified the mini prepped material. We then ran restriction digest(SpeI, BSA, Buffer 4) of the mini prepped sample.

- We streaked plates from the overnight cultures of transformed bacteria (psB1A3, pSB1AT3, pSB1C3, pSB 1k3) and placed them into the 37C incubator.
- We prepared a presentation of our project for Wellesley college, whom we will be collaborating with during the summer :)
- We had a meeting with professor Doug and other undergrads from CIDAR lab.
June
Week 2
- Attended meeting with Wellesley team. Collaboration has started!
-
We performed PCR amplification (20 μL reactions) with new sets of L0 primers: DVL0_EF with DVL0_BR (MS8), DVL0_FF with DVL0_BR (MS9), DVL0_GF with DVL0_BR (MS10), DVL0_DF with DVL0_FR (MS11), DVL0_DF with DVL0_GR (MS12) and DVL0_DF with DVL0_HR (MS13). We then performed gel electrophoresis of MS8-13, all were successful.
- We performed Gel extraction and nanodropped the samples.
- Ran 50 μL PCR reactions of MS8.1-13.1, the same samples as MS8-3, in order to have enough DNA for restriction digest. We performed Gel Electrophoresis of the PCR samples and we performed Gel Extraction. Then, we nanodropped the samples.
- We performed Restriction digest with the samples MS9.1 and MS 12.1 that had enough DNA concentration. We PCR clean up-ed MS9.1 and 12.1




-
We performed a new PCR in triplicate for the samples MS8.1, 10.1, 11.1 and 13.1 to achive a higher concentration of DNA. We ran gel of MS8.1, 10.1, 13.1,11.1 to amplify more DNA template to achieve sufficient DNA concentration upon gel extraction
- We performed ligation of the successful amplified samples of MS4-7 and MF4: MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3 and MF4 with backbone of 1K3


- Ran MgCl2 gradient of MF3 PCR to troubleshoot, along with DMSO separately.
- We transformed the ligations:MS4-7 with backbone of 1AT3, MS4-7 with backbone of 1A3, MF4 with backbone of 1K3 with competent bacterial cells. We spread plated them with 20 μL of a 40 mg/mL X-Gal solution and 8 μL of a 0.5 IPTG solution for blue-white screening.
June
Week 3
- We found out that MS8-13 are all wrong due to primer dimer. We redid the PCR of all level 0 primers that failed: L0AB (FJ1), L0EB (FJ2), L0FB (FJ3), L0GB (FJ4), L0DF (FJ5), L0DG (FJ6),L0DH (FJ7), L0DE (FJ8), L0CD (FJ9). Instead of running a gel after the PCR, PCR clean up was performed. The product of the clean up was then nanodropped.

-
Then the PCR clean up samples of FJ1-9 underwent restriction digest. We ran gel of digest, bands came out too weak, but all present.

-
Redid digest with higher amount of DNA. New gel is much more clear

-
We perfomed gel extraction and nanodropped gel extraction product.

-
Performed PCR clean up on RD of 1A3, 1AT3, 1C3, 1K3 transformed at week1 and nanodropped

-
Ligated 1K3 with pSMJSAF from samples of FJ1-9. Transformed the ligations on to plates. The plates did not yield positives for the transformation.
-
RD the backbones in two sets, one set with just SpeI other with both SpeI and EcoRI. Gel of the RD showed that there were unexpected bands when cut with both enzymes, something not shown on the registry experience pages for the backbones.

-
Plated bacteria transformed with the backbones. Picked colonies from the plates. Miniprepped and nanodropped the cultures with the the backbone

-
Made glycerol stocks of 1AT3, 1A3, 1C3, 1K3 both for -20 and -80 celsius
-
We prepared plate media of Kan, Amp and CAM
June
Week 4
-
We worked on Safety Answers for our Wiki.
-
We attended an Eugene Tutorial given by Ernst, the Pos-Doc responsible for Eugene in CIDAR Lab.
-
We worked on Human Practices project.
-
We met the Wellesley and MIT iGEM teams for a Physics lecture in MIT given by Walter Levine, had lunch together and visited the MIT iGEM team lab :)
We designed primers and gene synthesis template.
July
Week 1 - Happy July 4th!
-
We transformed plasmids containing the genes, promoters and terminator to be used as templates for the primers that we had designed. The plasmids come from the iGEM kit plates.
- we used bioline cells but didn’t follow the protocols because the cells were more competent so didn’t need to go through heat shock.
- cells sat on bench over the weekend. Colonies will be picked next week!
- We also autoclaved Broth, Media and DI water to have enough plates for our next round of transformations.
- We used to Clotho to input the primers that we had designed into a database.
- in doing so, we learned how to use Spreadit Features, Spreadit Oligos, Registry Importer, Collection Viewer and Sequence Viewer.
- We are still waiting for our primers to continue wet lab work.
July
Week 2
- We transformed the following parts from the iGEM plates into bioline cells : E1010, C0051, C0040, B0015, I13458, E0030, E0040, C0012, C0071, R0010, R0063, R0079, I13453, C0079, C0080, R0040, R0051, R0062, R0071, C0062. Cultures from transformation were made and then a miniprep was performed on all of them. We ran a gel to confirm the plasmid presence and performed nanodrop to measure DNA concentration. After that, glycerol stocks were made for all of them.
- We created working and stock solutions for the new primers that arrived to use at PCR. We performed PCR using the transformed parts as templates and the primers we designed in order to add moclo fusion sites to them. The PCR conditions were set according to the table below.

- We confirmed the PCR reactions by running 5μL agarose gels. For samples bigger than 300bp we ran 1% gel, for samples smaller than that we ran 2% gels.






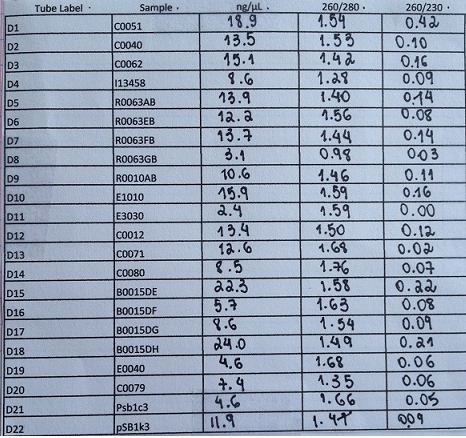
- We performed PCR Clean up in the samples: C0051, C0040, C0062, I13458, R0063AB, R0063EB, R0063FB, R0063GB, R0010AB, E1010, E3030, C0012, C0071, C0080, B0015DE, B0015DF, B0015DG, B0015DH, E0040, C0079, pSB1C3, pSB1k3. Then, we nanodropped them.

July
Week 3
-
We performed restriction digest of the samples with SpeI and NEB buffer 4 for 1 hour. Then, we performed PCR clean up of the RD samples and measured the DNA concentration by nanodrop.
- We did ligation of pSB1C3 and pSB1k3, in order to circularize the plasmid that we amplified in order to remove RFP.Then, we transformed the ligation product using bioline cells and spread plate them. The 1C3 yielded colonies but the 1K3 didn’t. We picked colonies of 1C3 for overnight culture.
- We performed PCR on pSB1A3, R0010(EB, FB, GB) and R0079 (AB, EB, FB, GB) with phusion from NEB to trouble shoot. For PCR we extended 1A3 extension time, while lowering annealing temperature for all samples. We run a gel to check the PCR product. For 1A3, we ran a 1% gel and for the other samples we ran a 2% gel. Again the amplifications did not work and we decided to double check the primers we designed. We realized that the primers R0010_For_E, R0010_For_F, R0010_For_G, R0079_For_A, R0079_For_E, R0079_For_F an R0079_For_G had problems on their design and couldn`t anneal properly to the template. We are ordering new correct primers and troubleshooting the amplification of pSB1A3.

- We performed Miniprep in sample pSB1C3 and digested it with SpeI and buffer N4. For the sample pSB1k3 we performed a new ligation, transformed the ligation product using bioline cell and spread plate it. We ran a 1% gel to check the digestion product of pSB1C3 and proceeded to PCR clean up of the sample and DNA quantification.


- We started building our level 0 destination vectors :) We ligated our backbone pSB1C3 with the following pcr amplified products: DVL0_GF_BR, DVL0_DF_FR, DVL0_DF_GR, DVL0_CF_DR and DVL0_BF_CR. We transformed them using Bioline competent cells, and plated them with X-gal and IPTG for blue-white screening.
- We updated our Clotho experience section, check it out!






















 .
.












