 "
"
Team:Paris Bettencourt/Restriction Enzyme
From 2012.igem.org
(→Characterisation of pRha) |
(→Characterisation of pRha) |
||
| Line 227: | Line 227: | ||
|[[Image:Paris_Bettencourt_2012_RG_pRha_photo_2.jpg|thumb|250px|center|<font size="1"><i>E.Coli</i> The same tubes under excitation light (540nm). Photo is taken through emission filter (590nm). We can clearly see that <b>the right tube</b> tube which was induced by L-rhamnose express RFP, while <b>the left tube</b> where we didn't add it has no expression.</font>]] | |[[Image:Paris_Bettencourt_2012_RG_pRha_photo_2.jpg|thumb|250px|center|<font size="1"><i>E.Coli</i> The same tubes under excitation light (540nm). Photo is taken through emission filter (590nm). We can clearly see that <b>the right tube</b> tube which was induced by L-rhamnose express RFP, while <b>the left tube</b> where we didn't add it has no expression.</font>]] | ||
|- | |- | ||
| - | |[[Image:Paris_Bettencourt_2012_RG_pRha_graph_2.jpg|thumb|550px|center|<font size="1">Hi</font>]] | + | | rowspan = 2|[[Image:Paris_Bettencourt_2012_RG_pRha_graph_2.jpg|thumb|550px|center|<font size="1">Hi</font>]] |
|} | |} | ||
Revision as of 18:48, 26 September 2012

- Main
- Team
- Project
- Achievements
- Human Practice
- Safety
- Notebook
- Attributions
 Overview
Overview Delay system
Delay system Semantic containment
Semantic containment Restriction enzyme system
Restriction enzyme system MAGE
MAGE Suicide system
Suicide system Encapsulation
Encapsulation Synthetic import domain
Synthetic import domain Safety Questions
Safety Questions Safety Assessment
Safety Assessment|
Achievements :
|
Contents |
Overview
Our group was responsible for designing a plasmid self-digestion system. This synthetic system allows to digest plasmids into linear parts of DNA and thus disrupt the expression of the genes carried by this plasmid, including the antitoxin (Colicin immunity protein), and any plugged-in synthetic device. Afterwards the cell's DNA can be degraded by the Colicin.
Objectives
- Find appropriate restriction enzymes which have to match the following properties:
- The corresponding restriction site must not be found in the E.Coli genome;
- The enzyme has to have high specifity;
- It has to work in wide range of different conditions (pH, T°, etc)
- Choose a strong yet tightly repressible promoter to regulate the restriction enzyme expression;
- Clone circuits with different combinations of the chosen restriction enzymes and promoters;
- Measure the degradation efficiency of the restriction enzyme for each circuit;
- Based on the best combination, design a self-disruption plasmid.
Design
According to the first two of our objectives, we should find an appropriate restriction enzyme and to choose a strong yet tightly repressible promoter to regulate its expression.
Restriction enzyme candidates:
- Fse I is a restriction endonuclease which recognizes an 8bp long DNA sequence: GGCCGG▽CC (CC△GGCCGG). The reason why we chose it is because it has the lowest number of restriction sites in the E.coli genome: only 4 copies. We decided to use MAGE to remove those sites from the chromosome (see for more details). However, MAGE did not have the expected yield, and we decided to freeze the work on this restriction enzyme and focus on the second candidate.
- I-SceI is an intron-encoded endonuclease. It is present in the mitochondria of Saccharomyces cerevisiae and recognises an 18-base pair sequence 5'-TAGGGATAA▽CAGGGTAAT-3' (3'-ATCCC△TATTGTCCCATTA-5') and leaves a 4 base pair 3' hydroxyl overhang. It is a rare cutting endonuclease. Statistically an 18-bp sequence will occur once in every 6.9*1010 base pairs or once in 20 human genomes.
Promoter candidates:
- pLac
We use a standard pLac promoter from Parts Registry: [] - pBad
- pRha L-rhamnose-inducible promoter is capable of high-level protein expression in the presence of L-rhamnose, it is also tightly regulated in the absence of L-rhamnose and by the addition of D-glucose.
- L-rhamnose is taken up by the RhaT transport system, converted to L-rhamnulose by an isomerase RhaA and then phosphorylated by a kinase RhaB. Subsequently, the resulting rhamnulose-1-phosphate is hydrolyzed by an aldolase RhaD into dihydroxyacetone phosphate, which is metabolized in glycolysis, and L-lactaldehyde. The latter can be oxidized into lactate under aerobic conditions and be reduced into L-1,2-propanediol under unaerobic conditions.
- The genes rhaBAD are organized in one operon which is controlled by the rhaPBAD promoter. This promoter is regulated by two activators, RhaS and RhaR, and the corresponding genes belong to one transcription unit which is located in opposite direction of rhaBAD. If L-rhamnose is available, RhaR binds to the rhaPRS promoter and activates the production of RhaR and RhaS. RhaS together with L-rhamnose in turn binds to the rhaPBAD and the rhaPT promoter and activates the transcription of the structural genes. However, for the application of the rhamnose expression system it is not necessary to express the regulatory proteins in larger quantities, because the amounts expressed from the chromosome are sufficient to activate transcription even on multi-copy plasmids. Therefore, only the rhaPBAD promoter has to be cloned upstream of the gene that is to be expressed. Full induction of rhaBAD transcription also requires binding of the CRP-cAMP complex, which is a key regulator of catabolite repression.
- The pRha sequence was containing an EcoRI restriction site, so we had to disrupt it in order to use pRha as a biobrick. In order to decide which base pair to modify, we used the [http://microbes.ucsc.edu UCSC Microbial Genome Browser]. We compared the pRha sequence in E.coli and similar species, and identified that the at the position is sometimes replaced by a in some species, so we decided to replace it in a same way. We ordered a gBlock with the pRha sequence having the mutation, and this is the sequence we used and submitted.
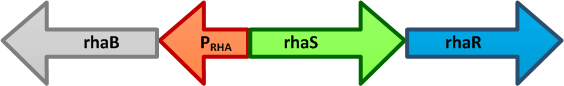 The E. coli rhaBRS locus. In the presence of Lrhamnose, RhaR activates transcription of rhaR and rhaS, resulting in an accumulation of RhaS. RhaS then acts as the L-rhamnose-dependent positive regulator of the rhaB promoter.
The E. coli rhaBRS locus. In the presence of Lrhamnose, RhaR activates transcription of rhaR and rhaS, resulting in an accumulation of RhaS. RhaS then acts as the L-rhamnose-dependent positive regulator of the rhaB promoter.

Considering these candidates, we decided to clone the following constructs in low-copy vector pSB3C5 to use it in our experiments, all with a medium RBS (B0032):
| pBad & RBS & I-SceI | pRha & RBS & GFP | pBad & RBS & RFP |
| pLac & RBS & I-SceI | pRha & RBS & GFP | pLac & RBS & RFP |
| pRha & RBS & I-SceI | pRha & RBS & GFP | pRha & RBS & RFP |
Experiments and results
Mesuring efficiency of I-SceI (Cloned parts)
To mesure digestion efficiency of I-SceI, we did a trasformation of two plasmids with different antibiotic resistance into NEB Turbo E.Coli strain:
- First plasmid: Low copy plasmid with encoded generator to express I-SceI meganucllease. Three version with different promoters was tested: I-SceI meganuclease controlled by pBad, pLac and pRha. For all version:
- Backbone: pSB3C5
- Resistance: Chloramphenicol
- Replication of Origin: modified pMB1 derived from pUC19
- Second plasmid: High copy plasmid with encoded I-SceI restriction site, [http://partsregistry.org/Part:BBa_K175027 K175027]. This biobrick was send us by TUDelft iGEM team.
- Backbone: pSB1AK3
- Resistance: Ampicillin and Kanamycin
- Replication of Origin: modified pMB1 derived from pUC19
As result we expected to select colonies with both plasmid, and after to measure efficiency of I-SceI after expression induction.
Transformation results
From the experiment we can clearly see that on plats with two antibiotics (Chloramphenicol & Ampicillin) there is no colonies, while on plates with only one antibiotic (Chloramphenicol or Ampicillin) there are numerous colonies.
 First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
We suggested two hipothesis to explain results:
- Two plasmids are not compatibles. Plasmids could have different origins of replication. That is why double transformation is unsuccessful.
- Our system works. Our system perfectly works, but there is some leakage expression of I-SceI meganuclease. In such case, it cuts I-SceI restriction site thus digest the second plasmid with ampicillin resistance.
Firstely, we decided to check the first hipothesis, and to check if two plasmids are compatible with each other.
Control for plasmid compatibility
As control experiment, we decided to trasform two plasmids into NEB Turbo E.Coli strain. The first plasmid in this experiment is analogous to one from the previous experiment with GFP insted of I-SceI meganuclease:
- First plasmid: Low copy plasmid with encoded generator to express GFP meganucllease. Only one version with pLac promoter was tested, because they all have the same backbone plasmid, and consequently the same replication prigin.
- Backbone: pSB3C5
- Resistance: Chloramphenicol
- Replication of Origin: modified pMB1 derived from pUC19
- Second plasmid: High copy plasmid with encoded I-SceI restriction site, [http://partsregistry.org/Part:BBa_K175027 K175027].
- Backbone: pSB1AK3
- Resistance: Ampicillin and Kanamycin
- Replication of Origin: modified pMB1 derived from pUC19
As result we expected to have colonies on both type of plates: firstly, on plates with one antibiotic (Chloramphenicol & Ampicillin), secondly, on plates with both antibiotics.
Our expectetions became true, so these two plasmids has compatible replication origins. That means, basically that our circuits work, but there is some leaky expression of I-SceI meganuclease.
 First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & GFP [Cm] Second plasmid: I-SceI restriction site [Amp] |
To avoid leakage, in the next experiment we tryied to recover cells after transformation and plate it in the presance of glucose.
Recovery in glucose
 First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pBad & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pRha & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
Results
Present your results
Mesuring of I-SceI efficiency (TUDelft parts)
In our experiments we also used two biobricks which were send to us by TUDelf iGEM team:
- [http://partsregistry.org/Part:BBa_K175027 BBa_K175027]
- [http://partsregistry.org/Part:BBa_K175041 BBa_K175041]
Experimental setup
Describe the experiment
 First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
 First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |  First plasmid: pLac & RBS & I-SceI [Cm] Second plasmid: I-SceI restriction site [Amp] |
Results
Present your results
Characterisation of pRha
 |  |
 |
Experimental setup
Describe the experiment
Results
Present your results
References
- Janise Meyertons Nelson et al., «Fsel, a new type II restriction endonuclease that recognizes the octanucleotide sequence 5′ GGCCGGCC 3′»
- Wernette C. M., «Structure and activity of the mitochondrial intron-encoded endonuclease, I-SceIV», Biochem Biophys Res Commun. 1998 Jul 9; 248(1):127-33.
- Yisheng Kang et al., «Systematic Mutagenesis of E.coli K-12 MG1655 ORFs»
- Jeanine M. Pennington, «On Spontaneous DNA Damage in Single Living Cells», Ph.D. thesis, Baylor College of Medicine, Houston (2006):
- Susan M. Rosenberg, «A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation»
- Colleaux et al., «Universal Code Equivalent of a Yeast Mitochondrial lntron Reading Frame Is Expressed into E. coli as a Specific Double Strand Endonuclease», (1986)