Saturday 30.06.12
Removed pBAD plates from 37 C incubator. Good growth on all transformed plates, Negative control showed no growth. Inoculated three colonies from plate B in liquid LB + Amp. Discarded the "mixed" plate. Placed plates A and B in refrigerator. Also inoculated three colonies from the lysis biobrick <partinfo>BBa_K112808</partinfo> using the plate from 22/6. Used an inoculating needle for all inoculations today.
Friday 29.06.12
LuxI (<partinfo>BBa_K092400</partinfo>) was transferred to petri dishes with Chloramphenicol and Kanamycin, to test if LuxI has a different resistance than given. A new batch of LA-medium with Ampicilin was prepared and poured on to petri dishes.
Researched the ArcA protein and its binding sequence(s) to investigate whether the lldPRD operon promoter can be modified to eliminate repression by ArcA-P in the anaerobic state. Located a putative ArcA-P binding site in the promoter sequence: The sequence GTTAACTAAATGTTA is the reverse complement of the minus strand of the 85 bp promoter sequence from position 38 to 52. This sequence is identified by McGuire et al (1999) (see this list). In the GenBank entry this part of the sequence reads TAACATTTAGTTAAC
Favorov et al. have made a newer suggestion for a general motif for ArcA binding sites - see this article and the computational result here. According to Favorov, the crucial features of the site is a direct repeat, as shown below:
atacaTAACatttagtTAACcattc
Extracted the pBAD strong promoter biobrick <partinfo>BBa_K206000</partinfo> and transformed it in two samples. Plated out on three Amp plates as follows: 200 uL from sample A, 200 uL from sample B, 20 uL each from sample A and B.
Thursday 28.06.12
A colony of LuxR (<partinfo>BBa_R0062</partinfo>) was transferred to liquid medium, but there were no colonies of LuxI (<partinfo>BBa_K092400</partinfo>). A new agar plate with LuxI was prepped.
Wednesday 27.06.12
Performed isolation of plasmid DNA from RBS (<partinfo>BBa_B0034</partinfo>), C78, C79 and VGF promoter (<partinfo>BBa_K561001</partinfo>). The parts coding for the LuxR and LuxI genes, <partinfo>BBa_R0062</partinfo> and <partinfo>BBa_K092400</partinfo> respectively, were transformed again.
Tuesday 26.06.12
Colonies from <partinfo>BBa_C0078</partinfo>, <partinfo>BBa_C0079</partinfo> and <partinfo>BBa_K561001</partinfo> were transferred from agar plates to liquid medium.
Performed isolation of plasmid DNA from the ribosome binding site (RBS) part <partinfo>BBa_B0034</partinfo>, and the double transcriptional terminator (DTT) part <partinfo>BBa_B0015</partinfo> grown on ampicillin and on kanamycin. The yield was quite poor, especially for RBS. We then decided to run the spin columns one more time to see if we could get a higher yield the second time. Since we are going to use a lot of RBS, we made a liquid culture right away, so we can perform a new DNA isolation on RBS tomorrow.
Wiki design and contents was revised. Front page layout done and general layout underway.
Monday 25.06.12
Research was done on hybrid promoters in the kit and two interesting ones were found. The genes coding for the necessary activators and repressors were also included in the distribution. The resulting three biobricks of interest <partinfo>BBa_K092400</partinfo> (LuxI), <partinfo>BBa_C0078</partinfo> (C78) and <partinfo>BBa_C0079</partinfo> (C79), were transformed using the standard protocol.
The BioBrick part <partinfo>BBa_K561001</partinfo> (vgb promoter, microaerobic) was extracted from the kit, transformed into E. coli, and the transformed cells plated out on Chloramphenicol (Cm) plates.
A transformed colony containing the RBS part extracted yesterday, was transferred to liquid medium (5 mL LB)
Two transformed colonies containing the DTT part on a plasmid backbone with Ampicillin and Kanamycin reistance, which was extracted yesterday, was transferred to liquid medium as follows:
One colony grown on agar plate with ampicillin, was transferred to a tube with LB (5 mL) + Kanamycin (100 ug/mL
One colony grown on agar plate with kanamycin, was transferred to a tube with LB (5 mL) + Ampicillin (100 ug/mL)
DNA was isolated from the liquid culture of LuxR+HSL transformed cells inoculated yesterday by miniprep. The DNA concentration in the product was measured as 13,3 ng/uL.
Sunday 24.06.12
The biobricks <partinfo>BBa_B0034</partinfo> (RBS) and <partinfo>BBa_B0015</partinfo> (Double terminator, DTT) were extracted from the distribution kit and transformed into E. coli which were plated out. The liquid culture left after the plating was stored at 5 C.
Plasmid DNA was isolated from liquid cultures, inoculated on 22.06 and 23.06 respectively, of E. coli transformed with <partinfo>BBa_R0062</partinfo> (LuxR & HSL promoter) and <partinfo>BBa_K112808</partinfo> (T4 lysis device, no promoter), using Promega miniprep. The concentration of isolated <partinfo>BBa_R0062</partinfo> DNA was measured to 15,7 ng/uL. Due to the low yield, this brick may have to be regrown. Suspecting too long incubation time as the cause. The concentration of isolated <partinfo>BBa_K112808</partinfo> DNA was measured to 111,0 ng/uL. Both samples were stored in the freezer at -20 C.
To make a new liquid culture of <partinfo>BBa_R0062</partinfo> transformed E. coli, a colony from the plate made on 21.06 (stored in refrigerator at 5 C since then) was inoculated in aproximately 4.5 mL LB medium with 4.5 uL of Amp stock solution (100 mg/mL) added, and incubated with shaking at 37 C.
Saturday 23.06.12
Of two LA + Amp plates with <partinfo>BBaK112808</partinfo> transformants (20 and 200 uL) incubated since yesterday, only the 200 uL plate showed growth (10+ colonies). Negative control plate (untransformed cells) showed no growth. One colony was transferred to liquid culture.
On the 20 uL plate, a water bubble below the agar was mistaken for a colony, and a liquid culture was inoculated with a toothpick after scratching the plate. The mistake was realized, but the tube was still left to incubate, as it was thought it could act as a (weak) negative control.
Friday 22.06.12
The BBa_R0062 transformant from yesterday yielded several colonies on its plate, and one colony was inoculated into liquid medium and incubated at 37 C with shaking. After adding antibiotic to the liquid medium in the growth tube, some of the medium was spilled, so the growth volume was about 2 mL.
Isolation of plasmid DNA from all 7 initial transformants (see 20.06.12) was performed with the Promega Wizard Plus SV Minipreps DNA Purification System A1460.
The biobrick <partinfo>BBa_K112808</partinfo> (Enterobacteria phage T4 Lysis Device - no promoter, called Lysis1) was extracted from the distribution kit and transformed into E. coli.
Thursday 21.06.12
We started the day with a lecture on genetic circuit modelling by Ph.d student and iGEM team instructor Marius Eidsaa, followed by a discussion of the options available in designing our system.
All 7 transformants from yesterday yielded 1 or more colonies. Negative controls (untransformed cells) plated out on Kanamycin and Ampicillin plates showed no growth, indicating that the antibiotics were effective in selecting transformants for growth. 1 colony from each of the transformants was inoculated in liquid LB medium and incubated at 37 C with shaking.
The biobrick BBa_R0062 (Promoter (luxR & HSL regulated -- lux pR)) was extracted from the distribution kit and transformed using our modified version of the official iGEM transformation protocol (see Protocols).
Wednesday 20.06.12
We started the day with an introduction to computer modeling of biological sytems, and use of the Cain chemical kinetics simulation program. We then researched and discussed various biobricks, and transformed several biobricks from the iGEM DNA distribution plates into E coli.
The following biobricks (with nicknames) were extracted from the iGEM 2012 DNA distribution kit and transformed according to protocol:
Tuesday 19.06.12
We started the day preparing lab equipment. We sterilized pipette tips, toothpicks, water and LA and LB medium. We also prepared stock solutions of ampicillin and kanamycin, and made petri dishes containing LA + Amp (100 ug/mL) and LA + Kan (100 ug/mL). Equipment and solutions were autoclaved at 120 C for 20 minutes.
We prepared LB medium (1 L) and LA medium (2 L). After mixing, each batch was divided into two bottles for autoclaving.
Stock solutions of Ampicillin and Kanamycin (both 100 mg/mL) were prepared from dry powders. After preparation and between use, the solutions were stored at -20.
After autoclaving, LA media bottles were left to cool. After reaching a temperature where they could be comfortably handled, the desired antibiotic was added using sterile technique. (500 uL stock solution to each bottle containing aproximiately 0.5 L, to a final antibiotic concentration of 100 ug/mL). The medium was then stirred and poured into petri dishes. The petri dished were left to cool down and placed cool for storage.
Monday 18.06.12
The team visited the lab and was given a EHS run-through by Merethe Christensen. We also risk evaluated the project and handed in the risk assessment, so now we are ready to start working in the lab:-)
Wednesday 13.06.12
Today, we tried to come up with a preliminary genetic circuit. We decided that using HGF as a signal molecule could be difficult, since we don't even know if a protein this size could penetrate the outer membrane and the peptidoglycan layer of E.coli. But we have found out that cancer cells excrete more lactate than healthy cells, so we decided to go for lactate, which is a small molecule. If the construct works, it could be modified to respond to other signal molecules.
Preliminary genetic construct:
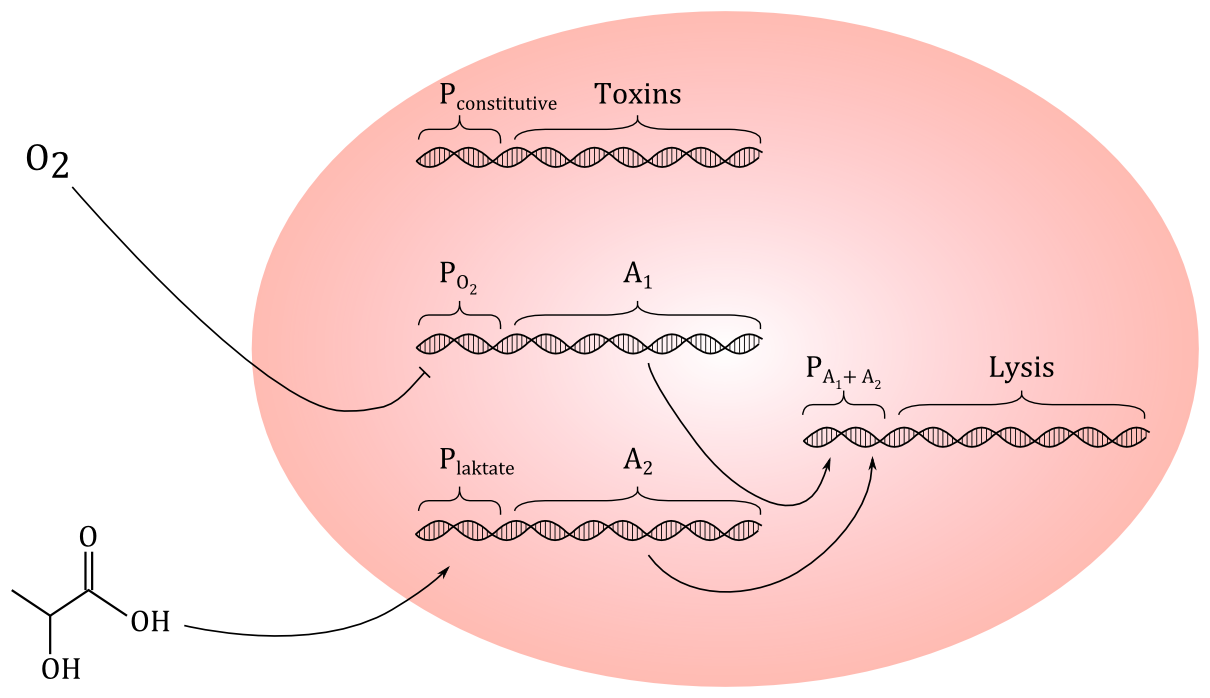
Preliminary sketch of the genetic circuit. A constitutive promoter will express toxins, while promoters regulated by O
2 and lactate will regulate the expression for proteins A
1 and A
2, which both needs to be present to start expression of the lysis genes
Friday 01.06.12
We talked about what we had found out since last week. Rolf and Jarle had since last week investigated different ways of making a cell lyse, and they found out that genes for lysis are already in this years iGEM kit.
Ove and Nina had been investigating toxins, but didn't find as much as they hoped for, but they have however found some toxins. Colisin E1 should be possible to use. They have also sent an email to Pål Fallnes, who Marit Otterlei suggested we could try to get in touch with. Apparently, he has been working quite a lot with expression of toxins in bacteria.
Gunvor and Eirin have since last week been trying to find a suitable signal molecule we could detect. Marit Otterlei suggested that we could use HGF, so Eirin and Gunvor did some reasearch around this growth factor. They found out that HGF is the only known lignad to the receptor c-Met, which is a tyrosin kinase. The idea so far is to use this, find out what signaling pathways this receptor is connected to, and find the endpoint of the signaling pathway. We assume that the endpoint is a growth factor possibly regulating a promoter, and if we find such a promoter, this could be set to control the lysis genes. The problem is that we don't know how signals are transducted to bacteria from the exterior, and this proved to be hard to find any information about. But we know that proteins resembling tyrosin kinases called BY kinases exists in bacteria, and also that proteins from eucaryotic cells have sometimes been working in bacteria when introns are removed by using mRNA and reverse transcriptase.
Both in the case of toxins we can express, and a signal molecule we could get our cells to respond to; we need more time. So we decided that Ove and Nina will continue looking for toxins for another week, and Eirin and Gunvor will investigate signal molecules more in depth.
Rahmi have found some possibly useful biobricks, and will be sending references to the biobricks in question to the team by email. Rahmi had also baked a cake for today's meeting:-)
In the case of sponsors, many of the companies we have asked request a budget. Gunvor will try to get hold of this from Eivind, and she will also talk to Merethe Christensen, who is an engineer at dept. of biotechnology, to arrange a safety excursion in the lab, which is necessary before we're allowed to work there.
We also elected Nina as our photo chief:-)
 "
"

