 "
"
Team:Peking/Modeling/Luminesensor/Optimization
From 2012.igem.org
| Line 57: | Line 57: | ||
</div> | </div> | ||
<p> | <p> | ||
| - | By searching the data of mutant in vivid protein among recent papers, we focus on these mutants: M135I in vivid dimerization domain to enhance K2 (vivid association equilibrium constant)<sup><a href="# | + | By searching the data of mutant in vivid protein among recent papers, we focus on these mutants: M135I in vivid dimerization domain to enhance K2 (vivid association equilibrium constant)<sup><a href="#ref3" title="Mechanism-based tuning of a LOV domain photoreceptor, Brian D. Zoltowski, etc. NATURE CHEMICAL BIOLOGY">[3]</a></sup> and I74V of amino acids surrounding Cys108 to enhance k1 (vivid decay rate constant)<sup><a href="#ref3" title="Mechanism-based tuning of a LOV domain photoreceptor, Brian D. Zoltowski, etc. NATURE CHEMICAL BIOLOGY">[3]</a></sup>. The experimental results validate our modeling prediction (See <a href="/Team:Peking/Project/Luminesensor/Characterization" title="">Characterization</a>). |
<br /> | <br /> | ||
</p> | </p> | ||
Revision as of 22:02, 25 September 2012








![]()
Parameter Analysis & Optimization
After modeling the origin system, we attempted to optimize it in a rational way. We have tuned the parameters both up and down, one by one, and finally discovered four parameters which predominantly influence this system.
| Function | Parameter | Description | Remark |
| Reduce responsing time | k1 | Vivid lighting decay rate constant | Mainly on process from Light to Dark |
| k3 | rate constant of monomer LexA releasing from specific binding site | ||
| Enhance contrast | K2 | Vivid association equilibrium constant | More dimerization provides more binding opportunity |
| K5 | dimered LexA binding equilibrium constant | More binding affinity |
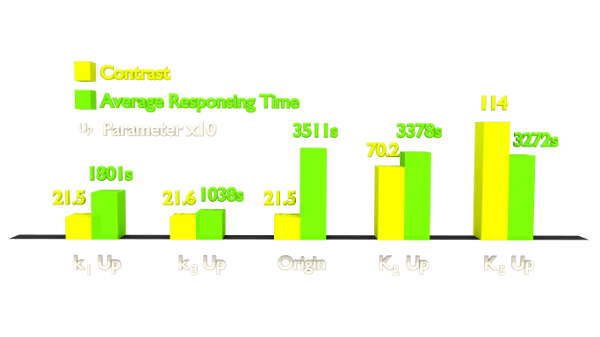
Figure 4. Results of Parameter Analysis. "Up" means tuning the parameter up to 10 times. This figure implied that tuning up these parameters can optimize the Luminesensor.
Within the two parameters to enhance contrast, K2 (vivid association equilibrium constant) is related to the association mechanism of Vivid protein and K5 (dimered LexA binding equilibrium constant) is related to the cooperative binding mechanism. As for speed optimizing, k1 (vivid decay rate constant) is related to the activation mechanism of Vivid protein and k3 (monomer LexA releasing rate constant from specific binding site) is related to the binding mechanism, thus also to the LexA and sequencing. If we change the binding affinity of the sequence, then it is difficult to make constant K3 (monomer LexA binding equilibrium constant with specific binding site), whose variance is predicted to ruin the contrast of this system from simulation. Therefore, we chose to apply a mutation on the Vivid protein in order to attain a faster Luminesensor, which has high levels of k1 (vivid decay rate constant).

Figure 5. Molecular modeling of mutation I74V, for gaining a faster Luminesensor. We could see that I74V is in the vicinity of Cys108, in order to enhance k1 (vivid decay rate constant).

Figure 6. Molecular modeling of mutation M135I, for gaining a larger contrast (on/off ratio). We could see that M135I is in vivid dimerization domain, in order to increase K2 (vivid association equilibrium constant).
By searching the data of mutant in vivid protein among recent papers, we focus on these mutants: M135I in vivid dimerization domain to enhance K2 (vivid association equilibrium constant)[3] and I74V of amino acids surrounding Cys108 to enhance k1 (vivid decay rate constant)[3]. The experimental results validate our modeling prediction (See Characterization).
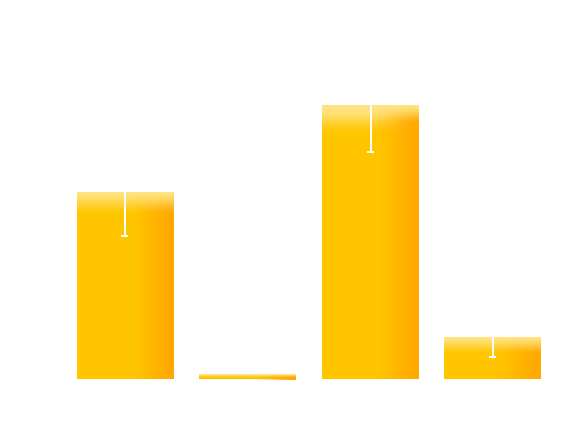
Figure 7. Experiment result: Effects of introduced mutations on the contrast (on/off ratio) of Luminesensor. We can see that the mutation on M135 obviously improves the contrast (on/off ratio) of Luminesensor, which validate our modeling prediction.
We also chose LexA408, the mutant of LexA, in order to increase K5 (dimered LexA binding equilibrium constant)[5], even though our main reason for choosing LexA408 over the wild-type LexA is due to the bio-orthogonality.
Figure 8. Molecular modeling of LexA408.



