 "
"
Team:Tianjin/Protocol
From 2012.igem.org
(Difference between revisions)
Austinamens (Talk | contribs) |
|||
| Line 162: | Line 162: | ||
## Sequence | ## Sequence | ||
| + | =DNA Assembler in Yeast= | ||
| + | ===SC Minimal Medium and Plates=== | ||
| + | SC is synthetic minimal defined medium for yeast. | ||
| + | 0.67% yeast nitrogen base (without amino acids) | ||
| + | 2% carbon source (i.e. glucose or raffinose) | ||
| + | 0.01% (adenine, arginine, cysteine, leuine, lysine, threonine, tryptophan, uracil) | ||
| + | 0.005% (aspartic acid, histidine, isoleucine, methionine, phenylalanine, proline, serine, tyrosine, valine) | ||
| + | 2% agar (for plates) | ||
| + | |||
| + | # Dissolve the following reagents in 900 ml deionized water (800 ml if preparing medium containing raffinose). Note: We make medium and plates as we need them and weigh out each amino acid. Many researchers prepare 100X solutions of each amino acid that they need.<br>'''Reminder: Omit uracil to make selective plates for growing pYES2 transformants.'''</br> | ||
| + | # If you are making plates, add the agar after dissolving the reagents above. | ||
| + | # Autoclave at 15 psi, 121°C for 20 minutes. | ||
| + | # Cool to 50°C and add 100 ml of filter-sterilized 20% glucose or 200 ml of filter-sterilized 10% raffinose. | ||
| + | # Pour plates and allow to harden. Invert the plates and store at 4°C. Plates are stable for 6 months. | ||
Revision as of 23:22, 23 September 2012




















Contents |
Gel Extraction Procedure
Excising and Dissolving the Gel
- Excise a minimal area of gel (up to 400 mg) containing the DNA fragment.
- Add Gel Solubilization Buffer (L1) to the excised gel in the tube size as indicated:
- Place the tube with the gel slice and Buffer L1 into a 50°C water bath or heat block. Incubate the tube at 50°C for 15 minutes. Invert the tube every 3 minutes to mix.
- Purify DNA using Centrifugation as described below.
Purifying DNA using Centrifugation
- Load. Place a column into a 2-mL Receiver Tube. Pipet the dissolved gel piece onto the column. Centrifuge the column at >12,000 × g for 1 minute. Discard the flow-through and place the column into the 2-mL Receiver Tube.
- Wash. Add 500 μL Wash Buffer (L2) containing ethanol to the column. Centrifuge the column at >12,000 × g for 1 minute. Discard the flow-through and place the column into the Receiver Tube. Centrifuge the column at maximum speed for 1 minute.
- Elute. Place the column into a clean 1.5 mL microcentrifuge tube. Add 50 μL of TE Buffer to the column. Incubate the tube for 1 minute at room temperature. Centrifuge the tube at >12,000 × g for 2 minutes.
- Store. The elution tube contains the purified DNA. Store the purified DNA at −4°C for immediate use or at−20°C for long-term storage.
Agarose Gel Electrophoresis
General Procedure
- Cast a gel
- Place it in gel box in running buffer
- Load samples
- Run the gel
- Image the gel
Casting Gels
The amount of agarose to use in your gel depends on the DNA in question. Use the following table as a rough guide:
- Measure out the appropriate mass of agarose into a beaker with the appropriate volume of buffer (see the documentation for your gelbox -- 50mL makes a good, thick gel for a 7x10cm gelbox).
- Microwave until the agarose is fully melted. This depends strongly on your microwave, but a 90 seconds at full power or 3 minutes at half power seem to provide decent results. As long as you do not burn the agarose and nothing bubbles over, this step is robust.
- Let the agarose cool on your bench until touching the bottom of the beaker with your bare hand doesn't burn you (~5 minutes for a 50mL gel).
- At this point add your DNA stain, e.g., ethidium bromide. The beaker will cool unevenly (surface first), so you must be careful not to cause ripples and bubbles.
- While the solution is cooling, seal the open edges of your gel box with one long piece of masking tape on each side. Make sure it is sealed well or the gel will leak.
- Pour the agarose solution into the taped gelbox. Carefully pop or shove to the side any bubbles, put in the comb, and let it cool for about 30 minutes, until the gel is solid.
- If your gel is at all purple, and you are using ethidium bromide as the DNA stain, you need to decrease your concentration by at least a factor of ten.
According to [http://openwetware.org OpenWetWare.org]
T4 Ligase Ligation
Materials
- T4 DNA Ligase
- 10x T4 DNA Ligase Buffer
- Deionized, sterile H2O
- Purified, linearized vector (likely in H2O or EB)
- Purified, linearized insert (likely in H2O or EB)
Procedure
10μl Ligation Mix
- 1.0 μL 10X T4 ligase buffer (use 10µl aliquots in -20 freezer; repeated freeze-thaw cycles can degrade the ATP in the buffer that's critical for the ligation rxn)
- 6:1 Molar ratio of insert to vector (~10ng vector). Gradients are used sometimes.
- Add (8.5 - vector and insert volume)μL ddH2O
- 0.5 μL T4 Ligase
Method
- Add appropriate amount of deionized H2O to sterile PCR tube
- Add 1 μL ligation buffer to the tube.
- Pipette buffer up and down before pipetting to ensure that it is well-mixed.
- Add 0.5 μL T4 ligase. PIPETTE half the volume of the mixture UP AND DOWN to ENSURE MIXING OF THE ENZYME.
- Also, the ligase, like most enzymes, is in some percentage of glycerol which tends to stick to the sides of your tip. Just touch your tip to the surface of the liquid when pipetting to ensure accurate volume transfer.
- Let the 10 μL solution incubate at 25°C for 15mins.
- Store at 4°C.
- (Use agarose gel electrophoresis to check sometimes)
- Transform into cell.
PCR Overlap Extension
Procedure
- Design Primers:
- These primers are like bridges between the two parts you want to assemble together.
- You will order two primers which are complements of one another.
- These primers will each have a 60°C Tm with one part and a 60°C Tm with the other part.
- The "end primers" will not have any complements and will likely only have restriction sites.
- "Extension PCR" PCR amplify the necessary fragments separately
- Use a proofreading polymerase enzyme.
- Use an annealing temp of 60°C.
- Clean up the product using a DNA column.
- "Overlap PCR" Use cleaned up fragments as template in a PCR reaction:
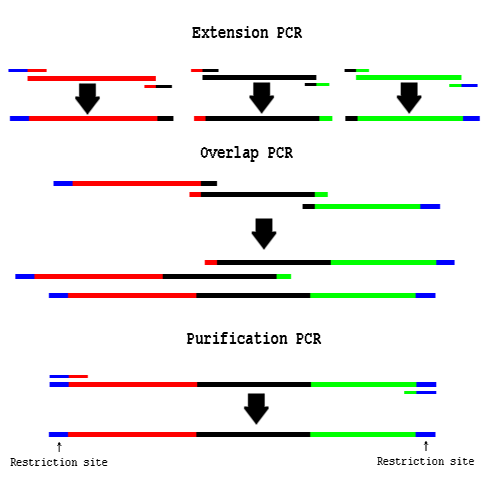 Diagram of PCR Overlap Extension
Diagram of PCR Overlap Extension
- About 1/2 to 3/4 volume of the Overlap PCR reaction should be equimolar amounts of purified fragments.
- Do not use Phusion polymerase. Try Pfu Turbo.
- Do not add any primers; the templates will prime each-other.
- Run 15 PCR cycles without primers.
- Use an annealing temp of 60°C.
- "Purification PCR" Add end primers to the Overlap PCR reaction:
- Continue cycling for another 15-20 rounds.
- Use an annealing temp of 72°C
- Gel extract the correct size fragment.
- Clone into the desired vector.
- Digest
- Ligate
- Transform
- Select
- Sequence

DNA Assembler in Yeast
SC Minimal Medium and Plates
SC is synthetic minimal defined medium for yeast. 0.67% yeast nitrogen base (without amino acids) 2% carbon source (i.e. glucose or raffinose) 0.01% (adenine, arginine, cysteine, leuine, lysine, threonine, tryptophan, uracil) 0.005% (aspartic acid, histidine, isoleucine, methionine, phenylalanine, proline, serine, tyrosine, valine) 2% agar (for plates)
- Dissolve the following reagents in 900 ml deionized water (800 ml if preparing medium containing raffinose). Note: We make medium and plates as we need them and weigh out each amino acid. Many researchers prepare 100X solutions of each amino acid that they need.
Reminder: Omit uracil to make selective plates for growing pYES2 transformants.</br> - If you are making plates, add the agar after dissolving the reagents above.
- Autoclave at 15 psi, 121°C for 20 minutes.
- Cool to 50°C and add 100 ml of filter-sterilized 20% glucose or 200 ml of filter-sterilized 10% raffinose.
- Pour plates and allow to harden. Invert the plates and store at 4°C. Plates are stable for 6 months.