|
|
| (4 intermediate revisions not shown) |
| Line 22: |
Line 22: |
| | | | |
| | </div><div id=note></html> | | </div><div id=note></html> |
| - | '''June 15'''
| + | [[File:Joe1Team_british_columbia_2012.png|centre|400x500px]] |
| - | | + | |
| - | A very busy day!
| + | |
| - | | + | |
| - | 1)Made 1 liter's worth of Kan plates, and about 3/4 liter's worth of chlor plates. We are now out of empty plates, and they have been placed on the list of things to buy. The Hallam lab is also out of empty plates. This is not good, for many of our knockouts are replaced by tetracycline resistance, and we have no tetracycline plates. We may have to scrounge up some from other labs. Noting that the kan plates that we used previously often grew colonies for negative controls, we increased the amount of kan stock we added from 1 mL to 1.5 mL. We have yet to test whether that is too great for the cells with the appropriate plasmid, but we have confirmed that it is lethal to normal cells by the fact that nothing grew on the negative test for the epi300 cells that Ting recently made.
| + | |
| - | | + | |
| - | 2)Tested epi300 cells by transforming with the rfp cassette for easy identification of whether or not the transformation was actually successful, or if other factors were in play regarding resistance. Negative controls were also employed for the newly made plates.
| + | |
| - | *Update: many colonies on the rfp plate, but none on the negative controls. The transformation efficiency appears to be slightly lower than previous competent cells, but is still fairly high.
| + | |
| - | | + | |
| - | 3)Miniprepped the resistance cassette colonies. Previously, to do the PCR, we just picked a colony, but we thought that it would be beneficial to have a plasmid stock in case we needed to repeat the PCR.
| + | |
| - | | + | |
| - | 4)PCR with new (biobrick) primers was attempted. We received primers for the Dsz genes and also for the amino acid genes. We did not, however, receive the DszA forward primer, and decided to hold off on working with the Dsz genes until later. We performed a cPCR for all the genes we had, and ran the results on the gel.
| + | |
| - | | + | |
| - | 5) We attempted to do a solvent tolerance assay, but were thwarted. As it turns out, gasoline has some rather unpleasant volatile compounds that were interpreted by other members of the LSC as dangerous. Also, the cells that we wanted to grow showed no growth for the first 5 hours. Personally, I think that while it would be nice to have the relevant data, it seems that the hurdles to test to see if the addition of the prefoldin chaperone helps our cultures grow in organic solvents to remove the sulfur compounds are too great. We may revisit the project, but right now, it seems that we should put the idea behind us. As of 2:18, June 16th, nothing is growing in John's other experiment, which may or not be documented on the wiki.
| + | |
| - | | + | |
| - | 6)Ran the gel. Below are the results from yesterday's and today's PCR. Cameron made the gel and taped the edges of the gel box to prevent the gel from escaping. The gel itself was made to 1% agarose in 0.5% TBE, and run on a 50 well gel box. 3 uL of loading buffer were used for 8 uL (10 uL for yesterday's products) sample to increase contrast, and various ladders were tested. The negative control for today's PCR popped open during the reaction, and evaporated. Despite this, I feel fairly confident that the data is accurate.
| + | |
| - | | + | |
| - | Running a gel
| + | |
| - | # Broad Range Ladder
| + | |
| - | # TrpA (biobricks)+DMSO
| + | |
| - | # TrpA
| + | |
| - | # TrpB +DMSO
| + | |
| - | # TrpB
| + | |
| - | # ArgC +DMSO
| + | |
| - | # ArgC
| + | |
| - | # ArgE +DMSO
| + | |
| - | # ArgE
| + | |
| - | # MetA +DMSO
| + | |
| - | # MetA
| + | |
| - | # TyrA +DMSO
| + | |
| - | # TyrA
| + | |
| - | # empty
| + | |
| - | # 1kb ladder
| + | |
| - | # Mehul's Stuff metA p1002 Kan
| + | |
| - | # Mehul's Stuff metA p1002 Kan
| + | |
| - | # Mehul's Stuff metA p1002 Kan
| + | |
| - | # Mehul's Stuff metA p1002 Kan
| + | |
| - | # empty
| + | |
| - | # Ruichen's Stuff
| + | |
| - | # Ruichen's Stuff
| + | |
| - | # Ruichen's Stuff
| + | |
| - | # Ruichen's Stuff
| + | |
| - | # Ruichen's Stuff
| + | |
| - | # empty
| + | |
| - | # 1kb + Ladder
| + | |
| - | # TrpA tet
| + | |
| - | # TrpA tet +DMSO
| + | |
| - | # TrpA tet +MgCl2
| + | |
| - | # TrpA tet +DMSO+MgCl2
| + | |
| - | # ArgE tet
| + | |
| - | # ArgE tet +DMSO
| + | |
| - | # ArgE tet +MgCl2
| + | |
| - | # ArgE tet +DMSO+MgCl2
| + | |
| - | # TyrA tet
| + | |
| - | # TyrA tet +DMSO
| + | |
| - | # TyrA tet +MgCl2
| + | |
| - | # TyrA tet +DMSO+MgCl2
| + | |
| - | | + | |
| - | [[File:June15igemmyriad.png |900x1000px]] | + | |
| - | * Figure 1. Myriad Gel
| + | |
| - | | + | |
| - | As you can see, we have successful bands for tyrA (from genome, biobrick primers) with DMSO, MetA with kanamycin resistance (for knockouts), trpA with tetracycline resistance, ArgE with tetracycline resistance, and tyrA with tetracyline resistance. The 1 kb+ ladder appears the most useful in this experiment.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | As for the organic solvent tolerance assay, indeed there has been no growing sign in the transformed culture (with 3I plasmid) for the first few hours, and it was left in the 37 degree room over night.
| + | |
| - | | + | |
| - | and for the controlled cultures, they were growing as expected when the organic solvents (Pentane and Octane) were not introduced, the experiment was continued (with the disruption of Jacob mentioned before). The organic solvent were added into controlled cultures at different concentrations (0%, 5%, 10%, and 15%), and their growth were monitored roughly each hour for the first few hours, and when the disruption occurred the cultures were taken out of the 37 degree room and stored in the 4 degree room at 5 p.m.
| + | |
| - | | + | |
| - | I returned to the Lab after dinner at 8:30 p.m., and each culture were examined for OD readings again, and the cultures with Pentane and Octane were incubated in the incubator with vacuums at 37 degree for 1 hour and 30 minutes respectively. Then the OD were read again at 9:30 p.m..
| + | |
| - | | + | |
| - | [[File:British_Columbia_Octane Tolerance_Control.png|900x1000px]]
| + | |
| - | *Figure 2. Octane Tolerance Control Test
| + | |
| - | | + | |
| - | [[File:British_Columbia_Pentane Tolerance_Control.png|900x1000px]]
| + | |
| - | *Figure 3. Pentane Tolerance Control Test
| + | |
| - | | + | |
| - | - [[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | The Amp plates made the day before worked. However, the Kan plates (negative control, EPI300 pIJ790, and DH5a pIJ790) showed unexpected results. The negative control, which only had untransformed K12 cells plated, had more colonies growing than those from the plates with resistance-transformed EPI300 pIJ790 and DH5a pIJ790. Even then, there were only a few small colonies on the EPI300 pIJ790 plate and none at all on the DH5a pIJ790 plate. It looks like the Kan plates may not be working, and it is possible that the EPI300 and DH5a cells with the pIJ790 plasmid are not competent.
| + | |
| - | | + | |
| - | - [[User:Tingchiawong|Tingchiawong]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 16'''
| + | |
| - | | + | |
| - | The time I came in Joe was making the PCR gel.
| + | |
| - | | + | |
| - | The cultures with 3I plasmid that left for growth in the 37 degree room actually did turn a bit cloudy (6:00 p.m.).
| + | |
| - | | + | |
| - | Their ODs were then measured with a blank LB for reference, and the detailed info will be updated.
| + | |
| - | | + | |
| - | For the remaining cultures, 100µl of each culture was tested on Chlor plates with an control of K12 (20µl) to see if they are actually the cell cultures that of our interest, and also testing the chlo plates that are newly made, and the remaining cultures were stored in the 4 degree room for potential future usage.
| + | |
| - | | + | |
| - | The plates were left in the 37 degree room, and will be collected by Joe and stored in the 4 degree room.
| + | |
| - | | + | |
| - | - [[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 17'''
| + | |
| - | | + | |
| - | As shown above in the gel image, there is still some PCR that needs to be done. Additionally, it appears that Joe's knockout PCR was not working either, so that PCR was to be repeated as well. It appears apparent that increased concentrations of DMSO and magnesium chloride help the reaction, so the appropriate amount of each was added to the mastermix. The reactions were as follows: TrpB with Kan cassette, TrpA, TrpB, ArgC, and MetA.
| + | |
| - | | + | |
| - | Since we were planning to put these into biobricks, it was imperative that we made sure that there were no restriction sites. To do this, we used the NCBI data for the gene sequence and ran it through Nebcutter, searching for all illegal sites. No illegal sites were found in TrpB, TyrA, TrpA, ArgC, or MetA. Lucky.
| + | |
| - | | + | |
| - | As shown above, a reaction for the tyrA gene worked. Using this PCR product and the rfp plasmid, we digested with EcoRI and PstI and ligated with T4 DNA ligase. The resultant product was used to transform Epi300 cells. Tomorrow, we will check for white colonies among the red ones, which would suggest that the ligation was successful. A colony PCR will be done to confirm.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 18'''
| + | |
| - | Yesterday's plates did not grow, but the experiment was repeated today.
| + | |
| - | | + | |
| - | 1) Made tetracycline plates using LB agar and Hallam lab tetracycline, diluted 400 uL in 400 mL. Previous kan and amp plates were shown to not have LB in them, and were also discovered not to support growth. However, a working kan plate was gifted by Cameron and used for a knockout experiment.
| + | |
| - | | + | |
| - | 2) A gel from yesterday's PCR was shown to have some products that worked, while others did not. The MetA, TrpA, TrpB, and ArgE biobrick PCR's worked fine, but the ArgE-Kan cassette and the ArgC biobrick PCRs did not, and were not repeated today. From personal correspondence with Joe, his PCR was not working either, despite trying different cycles and conditions. He suspects that there may be something wrong with the template, given the inconsistent performance of the Kan plates.
| + | |
| - | | + | |
| - | 3) The biobrick PCRs that did work were digested with EcoRI and PstI, as was the psb1C3 linearized plasmid backbone.
| + | |
| - | it was unknown whether or not the plasmid had any methylation, so DpnI was used only in the PCR product digestion. The procedure will be uploaded to the wiki in the near future. A gel was made showing the ligation products.
| + | |
| - | | + | |
| - | 4) The biobrick ligation products were used to transform K12 cells and were plated appropriately. The successful amino acid-antibiotic cassettes were also transformed into EPI300, DH5a, and BL21 cells with the appropriate recombineering plasmid, and plated. To date, all the tet resistance strains have been plated, as well as Mehul's Amp resistant strain. The kanamycin resistant strains remain recalcitrant, perhaps due to, again, the inconsistent performance of the kanamycin plates.
| + | |
| - | | + | |
| - | 5) Joe, looking into rhodococcus growth, decided to make some TB.
| + | |
| - | | + | |
| - | It should be noted that several of the transformations sparked during the electroporation procedure. Some success has been reported with sparked cultures, so they were plated anyways.
| + | |
| - | | + | |
| - | As an update, here is a table showing what has been done so far.
| + | |
| - | | + | |
| - | {| class="wikitable"
| + | |
| - | |-
| + | |
| - | | Gene||Type||PCR||Ligation||Transformation
| + | |
| - | |-
| + | |
| - | |TrpA+Tet resistance||Recombineering||Successful||NA||?
| + | |
| - | |-
| + | |
| - | |TrpB+Kan resistance||Recombineering||Unsuccessful||NA||No
| + | |
| - | |-
| + | |
| - | |ArgE+Tet resistance||Recombineering||Successful||NA||?
| + | |
| - | |-
| + | |
| - | |ArgC+Kan resistance||Recombineering||Unsuccessful||NA||No
| + | |
| - | |-
| + | |
| - | |TyrA+Tet resistance||Recombineering||Successful||NA||?
| + | |
| - | |-
| + | |
| - | |MetA+Amp resistance||Recombineering||Successful||NA||?
| + | |
| - | |-
| + | |
| - | |MetA||Biobrick||Successful||Done||?
| + | |
| - | |-
| + | |
| - | |ArgE||Biobrick||Successful||Done||?
| + | |
| - | |-
| + | |
| - | |TrpA||Biobrick||Successful||Done||?
| + | |
| - | |-
| + | |
| - | |TrpB||Biobrick||Successful||Done||?
| + | |
| - | |-
| + | |
| - | |TyrA||Biobrick||Successful||Done||?
| + | |
| - | |-
| + | |
| - | |ArgC||Biobrick||Unsuccessful||No||No
| + | |
| - | |}
| + | |
| - | | + | |
| - | It should be noted that when a ligation has been done, it does not mean that it was successful. We will not know until we see growth on the plates and a cPCR of the colonies has been done. Even then, it would be useful to have it sequenced.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | Worked with Jacob on steps 3 and 4 listed above. I made the gel, practised loading samples into wells (with Ruichen), and learned that the machine used to see the results is finnicky. Unfortunately, the 1 kb reference ladder did not work (unexpectedly and for unknown reasons).
| + | |
| - | | + | |
| - | (Step 3) Transformation and plating of the biobrick PCR products were the last things we did today. Of the 5, only TrpB sparked during electroporation. 1 uL of PCR product was used to transform cells.
| + | |
| - | | + | |
| - | (Step 4) Of the successful amino acid/antibiotic cassettes, Jacob and I transformed genes "4", "7", "12" (Tet resistant) and "M3" (Amp resistant)- names of the genes to follow - into EPI300, DH5a, and BL21 all with the recombineering plasmid pIJ790. The ones that sparked during electroporation are: 4 (EPI300), 4 (DH5a), 7 (BL21), and M3 (BL21). Quite a mix, but the addition of DMSO and MgCl<sub>2</sub> to boost PCR efficiency increased the salt concentration of the solution and may have caused sparking. Gene 4 had both compounds added and 7 had DMSO (unsure about M3 - that was Mehul). Only 1 uL of PCR product was used to transform cells to reduce chances of sparking, which may have helped.
| + | |
| - | | + | |
| - | - [[User:Grace.yi|Grace.yi]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 19'''
| + | |
| - | | + | |
| - | Designed primers with Ruichen and Mehul.
| + | |
| - | | + | |
| - | More specifically, the primers were for constitutively expressed GFP, RFP, and YFP to be put in the PCC1FOS vector. An EcoRI site was added just before the start of the promoter, and a BamHI site was added to the 5' end of the reverse complement. The melting temperature was normalized to about 59°C, and the primers were checked for any secondary structure.
| + | |
| - | | + | |
| - | There were no colonies on the plates transformed with any putative biobricks, so the procedure was attempted again.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 20'''
| + | |
| - | | + | |
| - | Plated the cultures with 3I plasmid on Kan for control and Chlor for testing purpose.
| + | |
| - | | + | |
| - | - [[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | Still no biobrick colonies. Tried again with an altered ligation protocol. Also ordered a variety of kill switches from the registry, along with Joe's Panamanian Pseudomonas Rhamnosyltransferase. Repeated PCR on things that had previously failed, and got the final amino acid biobrick and Ruichen's knockout resistance construct. Joe's knockout resistance cassette remains recalcitrant. It should be noted that I ran reactions with either PFU or phusion, and only the phusion worked. This may have been because of the low volume (0.5 uL) used.
| + | |
| - | | + | |
| - | At the meeting, the gasoline debacle was discussed, as was the general progress of the project, and potential fixes for some of the problems that we have been having.
| + | |
| - | | + | |
| - | I suggested to Joe that he use the 2-17F biobrick for his forward kanamycin resistance cassette, and other ideas were thrown around as well.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | After consultation, we moved the IGTS8, IGTS9, and Rhodococcus JHV1 strains that were growing on terrific broth and also ones growing on LB from the 37C shaker to one at 30C.
| + | |
| - | | + | |
| - | - [[User:Tingchiawong|Tingchiawong]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 21'''
| + | |
| - | | + | |
| - | The culture has been proven to be transformed successfully with the 3I plasmid! They grew colonies on the Chlor plate and not the Kan plate.
| + | |
| - | | + | |
| - | Learned how to miniprep
| + | |
| - | | + | |
| - | Checked out some cool posters for the microbiology Conference, and found that there is a assay of interest. The assay (2,6 DCPIP) helps organisms to grow on a diesel contaminated soil.
| + | |
| - | | + | |
| - | - [[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | Today was relatively fruitful. All amino acids have been PCR'd out of the genome, all the resistance cassettes are ready for the knockouts, and several colonies appeared on the ArgE plate. One colony was selected and grown in culture. The rest of the biobrick ligations were redone and allowed to ligate overnight.
| + | |
| - | | + | |
| - | The digests of the products were done with EcoRI and PstI in buffer 3, as opposed to the EcoRI-HF and PstI in buffer 2 we did previously. This was done because PstI has 100% activity in buffer 3, as opposed to 75% activity in buffer 2. Also, we learned that EcoRI-HF has 0% activity in buffer 3.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | '''June 22'''
| + | |
| - | | + | |
| - | Transformed with the overnight (Room temperature) ligation, using 1 uL ligation mix and 1800V. Joe then plated the cells after recovery.
| + | |
| - | | + | |
| - | Cameron reported knockouts!
| + | |
| - | | + | |
| - | Found a MetA biobrick colony on the appropriate plate from 2 days ago. Decided to grow it up in culture and run a PCR to confirm that it was successful.
| + | |
| - | | + | |
| - | Had previously grown up 10 mL of each fluorescent strain (GFP, RFP, YFP), but the plate reader became unavailable.
| + | |
| - | | + | |
| - | Wanted to design mutagenesis primers, but couldn't remember what software to use. For future reference:
| + | |
| - | | + | |
| - | NCBI for FASTA
| + | |
| - | | + | |
| - | Nebcutter to find sites
| + | |
| - | | + | |
| - | Virtual Ribosome for associated peptide
| + | |
| - | | + | |
| - | Wikipedia for codon table
| + | |
| - | | + | |
| - | PrimerX for basic design.
| + | |
| - | | + | |
| - | To whom it may concern, I think doing 4 sequential site directed mutagenesis reactions on dszA is going to take far more time and nearly as much money as simply synthesizing it.
| + | |
| - | | + | |
| - | Joe appears to be successfully growing IGTS8 (Rhodococcus) and IGTS9 (Bacillus) in TB at 30°C, and Pseudomonas putida at 37°. He also reports being unsuccessful in doing a cPCR for the dszD gene.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | The IGTS8 and IGTS9 cell cultures in both terrific broth and LB grew dense and cloudy at 30C. Glycerol stocks will be made out of these.
| + | |
| - | | + | |
| - | We have been having issues with having no colonies grow on plates with cells transformed with PCR products ligated into vectors. One possible cause is the transformation itself. All of Joe's transformation resulted in a spark. He had added 5 uL ligated to competent cells. The high salt concentration may have heavily influenced the sparking. So, I redid some of the transformations in a smaller volume of ligate. I transformed Joe's ligated 3:3 dszB, 3:3 dszC, and 3:1 dszB plasmids into EPI300 cells. Added 0.5 uL ligate to 40 uL competent cells and electroporated them. The 3:1 dszB cells sparked during electroporation. The cells were left to recover undisturbed at 37C for an hour before plating 50 uL transformants onto Chlor plates.
| + | |
| - | | + | |
| - | - [[User:Tingchiawong|Tingchiawong]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 23'''
| + | |
| - | | + | |
| - | Made competent ''Pseudomonas putida'' cells using the [[Competent Cell Production]] protocol.
| + | |
| - | | + | |
| - | We had trouble getting the cells transformed with the ligated amino acid pathway genes to grow when plated. We're unsure if the problem lies in the digest, the ligation, or the transformation. Thus, we decided to redo all these steps.
| + | |
| - | | + | |
| - | Re-digested the "M," "TA," "TB," "TyrA," and "ArgC" PCR products provided by Jacob. Made a master mix of 5 uL NEB buffer 2, 0.5 uL BSA, 0.5 uL EcoRI HF, 0.5 uL PstI, and 18.5 uL dH2O. Incubated each of the mixtures of 4 uL master mix plus 4 uL PCR product in a Thermocycler for ____.
| + | |
| - | | + | |
| - | Re-ligated digest of "M," "TA," "TB," "TyrA," and "ArgC." We suspect that it may be the ligase we used that was at fault for the undesired results seen in previous experiments. So, we decided to add in more ligase and add in ligase from 2 different sources/tubes. We added 1 uL pSBIC3 EP, 4.0 uL dH2O, 1 uL T4 ligase buffer, and 0.5 uL from each tube of T4 ligase were added to 3 uL of the respective digest products. A control was also set up so that there was only plasmid DNA added. The ligate mixtures were placed in the Thermocycler to incubate at 16C for 30 minutes and inactive at 65C for 20 minutes.
| + | |
| - | | + | |
| - | John miniprepped MetA and ArgE, which are being prepared as potential biobricks.
| + | |
| - | | + | |
| - | A new set of PCR reactions was set up for MetA (miniprepped potential biobrick), TyrA (colony PCR), ArgE (miniprepped potential biobrick), and yddG (colony PCR). Made a ligation master mix by combining 60 uL Phusion 5X buffer, 147 uL dH2O, 15 uL DMSO, 12 uL MgCl2, and 18 uL 10 mM dNTP.
| + | |
| - | | + | |
| - | - [[User:Tingchiawong|Tingchiawong]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 25'''
| + | |
| - | | + | |
| - | Designed primers for SDM of dszB gene
| + | |
| - | | + | |
| - | PCRed yddG and Try A with Marianne
| + | |
| - | | + | |
| - | Had a Skype meeting with Calgary iGEM team
| + | |
| - | | + | |
| - | -[[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 26'''
| + | |
| - | | + | |
| - | Designed primers for SDM of dszC gene
| + | |
| - | | + | |
| - | Joined Jacob and Joe for transformation of Heat(chemical) competent cells:
| + | |
| - | | + | |
| - | {| class="wikitable"
| + | |
| - | |-
| + | |
| - | | Plasmid transformed||Volume of Ligation Mixture used (µL)
| + | |
| - | |-
| + | |
| - | |dszB, dszC (3 to 3 ratio, Old and New)||3
| + | |
| - | |-
| + | |
| - | |MetA, TrpA, TrpB, TryA, ArgC, positive and negative control||7
| + | |
| - | |}
| + | |
| - | | + | |
| - | -[[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 27'''
| + | |
| - | | + | |
| - | Run the gel of Try A and yddg genes at 95 Volts for 1 hr and 30 min, with 1Kb ladder.
| + | |
| - | | + | |
| - | -[[User:Ruichen|Ruichen]]
| + | |
| - | | + | |
| - | | + | |
| - | '''June 30'''
| + | |
| - | | + | |
| - | In the last few days, we have confirmed that our restriction enzymes work, that our electroshock cuvettes do not due to corrosion from bleach, and still have had no confirmed biobricks. We picked another candidate colony from the ArgE plate because the PCR of the plasmid between VF2 and VR (standard biobrick sequencing primers) was only about 200 bp, while we were expecting about 1.2 kb. We got the same result from our potential MetA colony. We received new ligase, and if that doesn't work, we are going to try the gibson assembly method. Using a previous restriction digest, we attempted another ligation of our new potential biobricks.
| + | |
| - | | + | |
| - | The first set of chemically competent cells did not grow anything. The procedure was repeated and completed today. The cells were transformed with a known plasmid and appropriately plated.
| + | |
| - | | + | |
| - | Rafael gave us 4 new electroshock cuvettes to temporarily replace the ones that we had destroyed. Joe used 3 of them to test his knockout, and as of today, there is one candidate colony growing on a 1/4 antibiotic concentration plate. A battery of tests have yet to be run to confirm that it is a knockout.
| + | |
| - | | + | |
| - | Yesterday, we went to Dr. Ramey, who had taught students who made a successful knockout using the lambda red system. They used a different recombineering plasmid than we did, and we received this plasmid. It was, however, growing in a strain that we were unfamiliar with, and we decided to isolate the plasmid and transform our Epi300 cells with it.
| + | |
| - | It is currently growing in a culture at 30°.
| + | |
| - | | + | |
| - | We received only the TrpB gene from the registry, and successfully grew it on a plate. We are now growing a culture to isolate the plasmid and if Joe's knockout is indeed a knockout, we can characterize the existing biobrick part by complementation.
| + | |
| - | | + | |
| - | We ordered the Gibson assembly kit, and it ran upwards of 700 dollars for 50 reactions.
| + | |
| - | | + | |
| - | Joe successfully PCR'd out the DszD gene from Rhodococcus genomic DNA that he isolated.
| + | |
| - | | + | |
| - | We made Tet, Kan, and Amp plates with 1/2 and 1/4 antibiotic concentration to try growing our knockouts. There was no growth from a resistance-less K12 cell on any of the plates tested.
| + | |
| - | | + | |
| - | Our PCR for the yddg and tyrA genes was unsuccessful.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''July 1'''
| + | |
| - | | + | |
| - | There are many (hundreds) of colonies on Joe's recombineering plates, and the concentration of colonies goes up as the concentration of antibiotic goes down. It appears that the addition of arabinose to the culture did nothing to increase recombineering efficiency. There are several large, well defined colonies on the full antibiotic plate. It appears that 2 days are required for sufficient growth at 30°.
| + | |
| - | | + | |
| - | With this success in mind, I PCR purified a couple of other knockouts (ArgE Tet and TrpB Kan) and transformed them, reusing the electroshock cuvettes we borrowed from Rafael. A pack of 50 was ordered so we could do more than 4 transformations per day. They are currently growing in the 30°C room on the second floor.
| + | |
| - | | + | |
| - | We only had one chlor plate left, and 4 potential biobricks which were to be put on the chloramphenicol resistant pSB1C3 plasmid. Since there was only one plate (and one electroshock cuvette), only the MetA biobrick ligation product was transformed.
| + | |
| - | | + | |
| - | The pKD46 recombineering vector that we received from Dr. Ramey grew well overnight at 30°C, and was miniprepped, and then transformed into the Epi300 strain. The recovery period was done at 30°C as well. If the transformation is successful, Ting will make electrocompetent cells out of the Epi300+pKD46.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''July 7'''
| + | |
| - | | + | |
| - | In the past couple of days, we have found candidate knockouts for all relevant genes but trpA, and attempted to confirm that they were all actual knockouts by PCR. Unfortunately, none of the PCR's worked. We suspect that it was because of a bad enzyme, so some of the reactions were repeated. Currently, the results are inconclusive as to whether or not the knockouts are actually knockouts. We also ordered the relevant knockouts from the Keio collection.
| + | |
| - | | + | |
| - | Colonies with potential biobricks of trpA, tyrA, argE, metA, and dszD were successfully grown. This time, chemically competent cells were used, and they were plated on 1/2 concentration chlor plates. We also transformed and grew out the previously characterized minC biobrick.
| + | |
| - | | + | |
| - | To physiologically characterize the knockouts, we made M9 plates, and plan to set up an amino acid gradient. Sadly, we ran out of M9 salts before we could make tet plates, so some knockouts can't be tested in this manner yet. We also made spec plates to grow some things from the registry, but a lawn grew on the negative control. Looking into the literature, it looks like the concentrated antibiotic we were using was not the correct strength.
| + | |
| - | | + | |
| - | PCR of the yddg gene failed yet again. It is possible that the primers are not correct.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''July 9'''
| + | |
| - | | + | |
| - | Does anybody else update the wiki these days?
| + | |
| - | | + | |
| - | We have been in contact with Calgary regarding kill switches. More details to follow later.
| + | |
| - | | + | |
| - | One of our potential knockouts and a positive control were grown on the M9 plates. There was growth on both plates, although the growth on the positive control was much faster and greater than the potential knockout. Since the potential knockout was able to grow on the amino acid-less M9 media, it appears to be not auxotrophic. Characterization with PCR continues to be difficult, and it was suggested that we simply sequence the strains.
| + | |
| - | | + | |
| - | The plates with the potential knockouts were placed in the 37° incubator overnight to cure the plasmid.
| + | |
| - | | + | |
| - | The potential biobricks were successfully miniprepped, and a PCR was performed to isolate what was actually between the VF2 and the VR primers in the plasmid. The product will then be run on a gel, and if it appears the correct length, will be sent for sequencing.
| + | |
| - | | + | |
| - | Our Pseudomonas competent cells appear to work (conferring antibiotic resistance), but they are not expressing rfp.
| + | |
| - | | + | |
| - | We made spec plates, but a lawn grew with our transformed cells. Interestingly, on top of this lawn grew several well defined colonies. We attempted to grow these colonies (and the proper negative controls) in varying amounts of spectinomycin. It is possible the plates had insufficient spec, or that the strain we are working with is naturally resistant (there was a lawn, but no large colonies, on the negative control).
| + | |
| - | | + | |
| - | Several more biobricks from the registry were transformed.
| + | |
| - | | + | |
| - | Joe made M9 plates with an amino acid, to further test his knockout.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| - | | + | |
| - | | + | |
| - | '''July 10'''
| + | |
| - | | + | |
| - | The PCR to isolate and amplify the insert from the potential biobrick showed many bands, characteristic of non-specific primer binding. As another test to see if the insert was correct, we cut with EcoRI and PstI to look for the appropriate insert size, and indeed found that there was an insert at about the right size. With this in mind, simply sequencing from the plasmid is the next step. However, our sequencing company only accepts orders until 1:30 PM, and it was 4:30 before everything that needed to be done was done.
| + | |
| - | | + | |
| - | The transformations from yesterday's transformations appeared to work. Also, interestingly, the colony from on top of the spec plate lawn, but not the spec plate lawn itself, was able to grow in 40 and 80 ug/mL spec.
| + | |
| - | | + | |
| - | Cameron reports knockouts from Yale.
| + | |
| - | | + | |
| - | - [[User:jacobtoth|Jacob Toth]]
| + | |
| | | | |
| | | | |
| | '''July 15''' | | '''July 15''' |
| - |
| |
| - | Sent the potential biobricks for sequencing last Friday, ordered primers for mutagenesis on DszC, ordered primers for attachment of fluorescent genes to amino acid genes, received all but yddg knockouts from Yale, found that the Arg knockouts work as described. Several other primers were also ordered, but I am not privy to their nature.
| |
| | | | |
| | An attempt at doing an experiment to calibrate the fluorescent proteins for the plate reader (looking for quenching, linear relationship between OD and fluorescence, etc) was done on Sunday, but as it turns out, we had the wrong promoter for a fluorescent protein, and the wrong antibiotic resistance for one of the other proteins. Given this, we gave up, and decided to do it later with the proper strains. | | An attempt at doing an experiment to calibrate the fluorescent proteins for the plate reader (looking for quenching, linear relationship between OD and fluorescence, etc) was done on Sunday, but as it turns out, we had the wrong promoter for a fluorescent protein, and the wrong antibiotic resistance for one of the other proteins. Given this, we gave up, and decided to do it later with the proper strains. |
| - |
| |
| - | Recently, there was a meeting with the kill switch team from Calgary. They suggested doing more sequencing, and that is indeed a good idea if we want to pursue this project. I think that the intein primers were ordered, but again, I am not privy to exactly what was ordered.
| |
| - |
| |
| - | Competent cells were made from each of the knockouts, and they worked well. The yddg strain was ordered through collaboration with Calgary as well as the keio collection proper in Japan.
| |
| - |
| |
| - | Today, I watched two pairs of eyes. One was black, and the other grey. And while the owners thereof, for the space of five seconds, walked past each other, the grey shot at the black, and the black fiddled the grey. Bonus points for identifying the poem without using a search engine.
| |
| - |
| |
| - | Our Gibson assembly kit came! Doing the math, it turns out that it is about 30 times more expensive than gold, so we will use it wisely. Personally, I would have liked to just order the enzymes and buffer reagents, but c'est la vie. Our biobrick assembly kit also came, but that is rather less exciting.
| |
| - |
| |
| - | The sequencing results from our amino acid biobricks came back, and it looks like only MetA was successful. It should be noted that the melting temperatures of the Vf2 and the VR primers are 60°, for cPCR validation of biobricks.
| |
| | | | |
| | Several new combinations of fluorescent genes, antibiotic resistances, and Keio strains were made. We used the wrong ones last weekend, but now, we are going to test TyrA- +GFP, TrpA- +RFP, MetA- +YFP, and also wildtype with the fluorescent genes. Recently, we switched backbones for the 13M RFP to Psb1C3, for we do not know the provenance of our current psb1C3 with rfp (we suspect it might be on the lac operon, as per the registry guidelines), and we also put the GFP on the psb1C3 instead of a kanamycin resistant plasmid, because we were transforming into kanamycin resistant auxotrophs. | | Several new combinations of fluorescent genes, antibiotic resistances, and Keio strains were made. We used the wrong ones last weekend, but now, we are going to test TyrA- +GFP, TrpA- +RFP, MetA- +YFP, and also wildtype with the fluorescent genes. Recently, we switched backbones for the 13M RFP to Psb1C3, for we do not know the provenance of our current psb1C3 with rfp (we suspect it might be on the lac operon, as per the registry guidelines), and we also put the GFP on the psb1C3 instead of a kanamycin resistant plasmid, because we were transforming into kanamycin resistant auxotrophs. |
| | | | |
| - | Just for qualitative fun, we are trying to grow the ArgC- and the ArgE- together in M9 to see if anything results, with negative controls for both. We also tried growing a wildtype with a TrpB auxotroph with rfp to look for qualitative red fluorescence. The fluorescence calibration experiment must be done before we can do these sorts of things quantitatively. | + | Just for qualitative fun, we are trying to grow the ArgC- and the ArgE- together in M9 to see if anything results, with negative controls for both. We also tried growing a wildtype with a TrpB auxotroph with rfp to look for qualitative red fluorescence. The fluorescence calibration experiment must be done before we can do these sorts of things quantitatively. |
| - | | + | |
| - | We miniprepped 4/6 of the biobricks that we ordered from the registry. We intend on sequencing them in the near future. We, lacking LB amp plates at the time, plated one on an M9 plate, but we now think that the strain the registry sent us is auxotrophic for something. We replated on LB amp after making new plates. The last one that we ordered did not arrive.
| + | |
| - | | + | |
| - | Joe made M9 plates with and without 1 mM arginine, and it was found that the two arginine auxotrophs grew as expected.
| + | |
| - | | + | |
| - | We bought a fridge for our new office, but have yet to fill it. If anyone with extra snacks reads this and wants to help, our fridge is currently underfilled.
| + | |
| - | | + | |
| - | Overnight cultures have been set up to make competent cells out of DH5a, K12, and IGTS8.
| + | |
| - | | + | |
| - | Some new polymerase system has found its way into our freezer, and nobody seems to know its provenance. Since we do not share the freezer, we assume some kindly benefactor gifted it to us.
| + | |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| - |
| |
| - |
| |
| - | '''July 17'''
| |
| - |
| |
| - | Today, we made dh5a and k12 competent cells. We also ran a cPCR on our biobrick colonies that turned out to not be biobricks.
| |
| - |
| |
| - | As said yesterday, we grew the two arg auxotrophs together, and they grew substantially more (~0.15 OD600) than the negative control. The wt grew to an OD of about 1. We are growing them for another night to see if the OD of the mix increases at all.
| |
| - |
| |
| - | Since sequencing confirmed a MetA biobrick, we transformed K12 cells with the remaining stock of our plasmid, so we could amplify it.
| |
| - |
| |
| - | All relevant transformed strains from yesterday grew well on the plates. We are now ready to properly do the fluorescence calibration experiment, but due to time constraints, we are going to do it on Thursday.
| |
| - |
| |
| - | - [[User:jacobtoth|Jacob Toth]]
| |
| - |
| |
| - | [[File:Pseudomonas Crystals.jpg|900x1000px]]
| |
| | | | |
| | | | |
| | '''July 19''' | | '''July 19''' |
| | | | |
| - | We tested the growth rate of our 3 main auxotrophs in various (log scale from 100 pM to 10 mM) concentrations of the amino acids to do monod kinetics. We also miniprepped our metA biobrick (so we have enough to send to the registry and use for experiments), new potential candidates for other biobricks (tyrA and ArgC), our construct which is identical to 13M on plate 3, except on a different plasmid, and a biobrick that we received from the registry. Sequencing to follow. | + | We tested the growth rate of our 3 main auxotrophs in various (log scale from 100 pM to 10 mM) concentrations of the amino acids to do monod kinetics, which will allow us to predict growth rates at various concentrations of the amino acids. |
| - | | + | |
| - | Joe has, to the best of my knowledge, identified potential biobrick colonies for the DszB,C, and D. Again, sequencing to follow.
| + | |
| - | | + | |
| - | We have, in the past few days, designed a plasmid that will aid in the testing of biobricks. The primers are to be designed and ordered today, if all goes according to plan. The primers that we tried to order through the lab a while ago spent some time in bureaucratic limbo, but I think they have been ordered now. Again, no one appears completely sure of what has been ordered, and one of our chief contacts on the subject has apparently gone on vacation.
| + | |
| - | | + | |
| - | It appears that Joe and Grace were attempting the fluorescence calibration experiment, but things apparently were going wrong, and we have not yet analyzed the data completely.
| + | |
| - | | + | |
| - | Concerning the kill switch plasmid parts, we want to take the psb1C3 plasmid, add a constitutively expressed LacI gene, and a lac promoter and rbs immediately before the cut sites. This way, a toxic protein can be well controlled. In fact, while we are designing this for toxic proteins, it could have uses in general protein production. Previous attempts for using the lac promoter on a high copy plasmid have failed leaky expression) due to the lack of the LacI protein produced in the genome. This new plasmid design remedies this, and will be accomplished by Gibson assembly.
| + | |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| Line 493: |
Line 63: |
| | Analyzed the data, and found out that the growth rate and the amino acid (Tyr) concentration has a correlation as follows: | | Analyzed the data, and found out that the growth rate and the amino acid (Tyr) concentration has a correlation as follows: |
| | | | |
| - | [[File:growth-rate at different try concentration_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different try concentration_british_columbia_2012.png|650px]] |
| | *Figure 4. cell growth-rate at different Tyr concentrations | | *Figure 4. cell growth-rate at different Tyr concentrations |
| | | | |
| - | [[File:growth-rate at different tyr concentration log scale_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different tyr concentration log scale_british_columbia_2012.png|650px]] |
| | | | |
| | *Figure 5. Log scale of cell growth-rate at different Tyr concentrations | | *Figure 5. Log scale of cell growth-rate at different Tyr concentrations |
| Line 509: |
Line 79: |
| | The growth rate and the amino acids (Met and Trp) concentration has a correlation as follows: | | The growth rate and the amino acids (Met and Trp) concentration has a correlation as follows: |
| | | | |
| - | [[File:growth-rate at different met concentration_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different met concentration_british_columbia_2012.png|650px]] |
| | | | |
| | *Figure 6. cell growth-rate at different Met concentrations | | *Figure 6. cell growth-rate at different Met concentrations |
| | | | |
| - | [[File:growth-rate at different Met concentration log scale_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different Met concentration log scale_british_columbia_2012.png|650px]] |
| | | | |
| | *Figure 7. Log scale of cell growth-rate at different Met concentrations | | *Figure 7. Log scale of cell growth-rate at different Met concentrations |
| | | | |
| - | [[File:growth-rate at different trp concentration_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different trp concentration_british_columbia_2012.png|650px]] |
| | | | |
| | *Figure 8. cell growth-rate at different Trp concentrations | | *Figure 8. cell growth-rate at different Trp concentrations |
| | | | |
| - | [[File:growth-rate at different trp concentration log scale_british_columbia_2012.png|900x1000px]] | + | [[File:growth-rate at different trp concentration log scale_british_columbia_2012.png|650px]] |
| | | | |
| | *Figure 9. Log scale of cell growth-rate at different Trp concentrations | | *Figure 9. Log scale of cell growth-rate at different Trp concentrations |
| Line 534: |
Line 104: |
| | Eventually arrived at the following graphs of growth rates observations, when n=3 | | Eventually arrived at the following graphs of growth rates observations, when n=3 |
| | | | |
| - | [[file: GrowthRatesAA_university_of_british_columbia.png|900x1000px]] | + | [[file: GrowthRatesAA_university_of_british_columbia.png|650px]] |
| | | | |
| - | [[file: GrowthRatesAA_LOG_university_of_british_columbia.png|900x1000px]] | + | [[file: GrowthRatesAA_LOG_university_of_british_columbia.png|650px]] |
| | | | |
| | From these graphs, the maximum growth rate, and Ks values that are used in Monod Kinetics can be determined. | | From these graphs, the maximum growth rate, and Ks values that are used in Monod Kinetics can be determined. |
| Line 544: |
Line 114: |
| | | | |
| | '''August 12''' | | '''August 12''' |
| - |
| |
| - | A large set of primers arrived, which gave us a lot to do.
| |
| | | | |
| | With a set of the primers that arrived, we were able to PCR amplify certain parts that will be able to be Gibson assembled. On the plasmid that has RFP (a modified BBa_K093012, placed on the Psb1C3), we wanted to put arabinose inducible MetA or arabinose inducible TyrA. However, one of the primers necessary to amplify the plasmid itself, which would be necessary to incorporate the proper homologous sequence for Gibson assembly, did not arrive. Additionally, the PCR to amplify the MetA gene did not work the first time, but has been repeated. The arabinose promoter was successfully PCR'd out of the E. coli genome for both cases. | | With a set of the primers that arrived, we were able to PCR amplify certain parts that will be able to be Gibson assembled. On the plasmid that has RFP (a modified BBa_K093012, placed on the Psb1C3), we wanted to put arabinose inducible MetA or arabinose inducible TyrA. However, one of the primers necessary to amplify the plasmid itself, which would be necessary to incorporate the proper homologous sequence for Gibson assembly, did not arrive. Additionally, the PCR to amplify the MetA gene did not work the first time, but has been repeated. The arabinose promoter was successfully PCR'd out of the E. coli genome for both cases. |
| Line 551: |
Line 119: |
| | We also wanted to put a IPTG inducible (BBa_K091111, LacIQ) metA gene after a GFP. The PCR for the MetA gene and the GFP plasmid were successful (although the GFP plasmid appeared as a fairly weak band on the gel), but the PCR for the LacIQ promoter failed. The initial DNA for this PCR was added directly from the parts distribution kit, and as such the composition and concentration of exactly what was added was unknown. The PCR was repeated as a cPCR on the genome, so as to at least get a usable part with the right overhang. | | We also wanted to put a IPTG inducible (BBa_K091111, LacIQ) metA gene after a GFP. The PCR for the MetA gene and the GFP plasmid were successful (although the GFP plasmid appeared as a fairly weak band on the gel), but the PCR for the LacIQ promoter failed. The initial DNA for this PCR was added directly from the parts distribution kit, and as such the composition and concentration of exactly what was added was unknown. The PCR was repeated as a cPCR on the genome, so as to at least get a usable part with the right overhang. |
| | | | |
| - | The final primers for the gibson assembly of the kill switch primer also arrived. There are four parts to this plasmid: the plasmid backbone, the lacI promoter, the constitutively expressed lacI gene, and the constitutively expressed RFP for easy selection. The plasmid backbone, the promoter, and the constitutively expressed RFP PCR products has been ready for several weeks, but we lacked the primers for the constituvely expressed lacI gene until now. To generate the DNA for the constitutively expressed lacI gene, we took BBa_K081005 (const. promoter+rbs) cut with only SpeI and BBa_I732100 (lacI) cut with only XbaI, and ligated the two together, and then did a PCR on the ligation. It worked beautifully.
| + | In an effort to see how much amino acid each type of cell in the co-culture will produce, we have been harvesting supernatants of our normal cultures as per the protocol in Kerner et al. Once we have supernatants of the cultures at various ODs, we are going to grow the auxotrophs in this supernatant, and then look at the growth rate and final OD. |
| - | | + | |
| - | The primers also arrived to isolate the VMA Sce-I intein, and Alina provided yeast genomic DNA. The PCR was done successfully. However, upon further reflection, it appears that we forgot to DpnI digest the original template for the plasmid backbone on which we were going to add this part, and we actually did not have any product for it. Another PCR needs to be done, and DpnI digests will be done before running it on a gel.
| + | |
| - | | + | |
| - | A previous potential part, ArgE, was restriction confirmed, and will be sent for sequencing at the soonest convenience.
| + | |
| - | | + | |
| - | The PCR for yddg was finally a success. The part has been restriction digested, along with the pSB1C3 plasmid, and a ligation will follow.
| + | |
| - | | + | |
| - | We finally received the DszA forward primer, and did a restriction digest on it as well. It will be ligated as well in the near future. It has several Pst1 sites, which limits some of the reactions we can do with it, but we can still place things before it by cutting with EcoRI and/or XbaI.
| + | |
| - | | + | |
| - | In an effort to see how much amino acid each type of cell in the co-culture will produce, we have been harvesting supernatants of our normal cultures as per the protocol in Kerner et al. Once we have supernatants of the cultures at various ODs, we are going to grow the auxotrophs in this supernatant, and then look at the growth rate and final OD. The list of which supernatants we have can be found in the lab. | + | |
| | | | |
| | Joe has found inconsistent results with his fluorescent proteins on the various promoters, and we have plans to standardize the promoter and rbs for the fluorescent proteins that will be used. Additionally, there is some overlap between YFP and GFP that is proving difficult to account for, so we are going to try to switch one to either BFP or CFP. | | Joe has found inconsistent results with his fluorescent proteins on the various promoters, and we have plans to standardize the promoter and rbs for the fluorescent proteins that will be used. Additionally, there is some overlap between YFP and GFP that is proving difficult to account for, so we are going to try to switch one to either BFP or CFP. |
| Line 573: |
Line 131: |
| | | | |
| | Having harvested the supernatant of the various cultures at various ODs, we filter sterilized it with a 0.2 uM filter to remove any cells that may have still remained. Now that we have the sterilized supernatant, we are going to follow the protocol in Kerner et al 2012, and add 1/5 5X M9 media, and grow different auxotrophic cultures, noting the OD and growth rate. From this, we will try to calculate, or at least approximate, the amino acid export rate of the cells. | | Having harvested the supernatant of the various cultures at various ODs, we filter sterilized it with a 0.2 uM filter to remove any cells that may have still remained. Now that we have the sterilized supernatant, we are going to follow the protocol in Kerner et al 2012, and add 1/5 5X M9 media, and grow different auxotrophic cultures, noting the OD and growth rate. From this, we will try to calculate, or at least approximate, the amino acid export rate of the cells. |
| - |
| |
| - | There were colonies on the MinC Intein plate, the ArgE BB plate, and the GFP construct LB plate, but nothing on the GFP construct M9 plate. This may have been because there was no lactose or IPTG to induce the metA gene. Interestingly, there was a green fluorescent patch, but it was rather weak and had no distinct colonies. We plan to pick several colonies from the LB plate and try growing them in M9 culture with and without IPTG overnight. We also plan to do a cPCR on the MinC plate using the intein primers. There were no colonies on the DszA or yddg plate. We have had no luck with electroporating cells with ligation mixtures, although we have had some success with heat shock competent cells. We think there may be something in the ligation mixture that is interfering with the electroporation, and could probably be fixed by purifying the ligation mix.
| |
| | | | |
| | The data that we collected regarding the growth curves of the auxotrophs and the mixture are very different from what the plate reader previously showed. We think this may be because of a lack of correction for path length by the plate reader, but aren't entirely sure. | | The data that we collected regarding the growth curves of the auxotrophs and the mixture are very different from what the plate reader previously showed. We think this may be because of a lack of correction for path length by the plate reader, but aren't entirely sure. |
| - |
| |
| - | We also did a PCR on the TyrA BB product, the previous failed YFP plasmid, and several other previously failed PCRs.
| |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| Line 586: |
Line 140: |
| | | | |
| | After two days of shaking at 37° in 1 mM IPTG, the supposed IPTG inducible metA constructs in the metA auxotrophs were unable to grow. | | After two days of shaking at 37° in 1 mM IPTG, the supposed IPTG inducible metA constructs in the metA auxotrophs were unable to grow. |
| - |
| |
| - | The gels of the PCR products that we have been getting lately have not been working properly. We think that there is a problem with the buffer due to how many times it has been reusued, or possibly by the new TBE that was recently made.
| |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| Line 596: |
Line 148: |
| | The supposed metA constructs still appears unable to complement a metA knockout. It has been suggested that the concentration of metA may have been too high, and this would have led to a severe detriment to the cell, and it has also been suggested that the IPTG, being a rather old stock, was no longer good. Another step that we are going to take is to see if the expression of the construct can be seen on an sds page under induction. | | The supposed metA constructs still appears unable to complement a metA knockout. It has been suggested that the concentration of metA may have been too high, and this would have led to a severe detriment to the cell, and it has also been suggested that the IPTG, being a rather old stock, was no longer good. Another step that we are going to take is to see if the expression of the construct can be seen on an sds page under induction. |
| | | | |
| - | Despite, or perhaps because of this failure, we are moving on, trying to make new constructs that will be used for tunability. The YFP has yet to give us a solid band, so we are trying to move it into the psb1c3 plasmid in case there are some plasmid specific sequences that were giving us poor results. The ligation and transformation has been done, and we are waiting for colonies. The PCR products necessary for the gibson assembly of the GFP-IPTG-TrpA gene were also all available, and that gibson assembly has been done. However, due to the failure of several gels, there was very little product left, and PCR purification would likely result in unusuable quantities, so we tried a Gibson assembly with the straight PCR product, and we will see if it works.
| + | Concnerning the other constructs, the PCR for the YFP has yet to give us a single solid band, so we are trying to move it into the psb1c3 plasmid in case there are some plasmid specific sequences that were giving us poor results. The ligation and transformation has been done, and we are waiting for colonies. The PCR products necessary for the gibson assembly of the GFP-IPTG-TrpA gene were also all available, and that gibson assembly has been done. However, due to the failure of several gels, there was very little product left, and PCR purification would likely result in unusuable quantities, so we tried a Gibson assembly with the straight PCR product. |
| | | | |
| - | All of the parts necessary for the assembly of the kill switch plasmid have been successfully PCR'd, but several of the parts fall victim to the same caveat as that of the previously discussed products. That being said, we tried it anyways,and will check for colonies tomorrow.
| + | Another PCR was done to generate some of the other parts required for assembly of other parts. The PCR for the arabinose promoter (using a new primer) has not yet resulted in a band. |
| | | | |
| - | The TBE that we were using for the gels has not been working for anyone in the lab, although it has been suggested that that may simply because people don't change their buffers. The TAE gels work fine.
| + | There are two distinct bands on the RFP PCR, one at the correct length, the other much lower. Several other products were also generated today, but there are still two essential parts, that of YFP and that of the new arabinose promoter, which have not yet been successfully PCR'd. |
| - | | + | |
| - | Another PCR was done to generate some of the other parts required for assembly of other parts. The new forward primer for the arabinose promoter does not seem to be working, although the old one worked well. We recently ran out of phusion polymerase, and have resorted to using an eclectic mix of collected polymerases in our PCR box.
| + | |
| - | | + | |
| - | A cPCR of the colonies with the MinC protein with the Sce VME intein showed that the gene for the intein was present in the cells that were supposed to carry it! A small victory, perhaps, but it shows that the Gibson assembly works, contrary to out experiences with the positive control. A site directed mutagenesis must be performed on the gene to convert an internal lysine into a phenylalanine, which has been shown to make the intein temperature sensitive. The primers for this reaction have been eluted, but we are still waiting for a plasmid stock of the minC-intein plasmid. By the way, the MinC protein was chosen because it can be used as a kill switch and has the necessary sequence (G-C) that the intein uses for self excision.
| + | |
| - | | + | |
| - | There are two distinct bands on the RFP PCR, one at the correct length, the other much lower. Seems like a perfect chance for gel extraction. Several other products were also generated today, but there are still two essential parts, that of YFP and that of the new arabinose promoter, which have not yet been successfully PCR'd. | + | |
| - | | + | |
| - | The cPCR on the DszA colonies gave no result. This may have been due to an unoptimized Taq protocol,and might be worth repeating.
| + | |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| Line 615: |
Line 159: |
| | '''August 17''' | | '''August 17''' |
| | | | |
| - | The Gibson assembled parts using unpurified PCR products did not work. I think it was because of the extra salts, etc that would be present in the PCR mix. As a note, make sure to somehow purify your PCR product before doing a Gibson assembly! | + | The Gibson assembled parts using unpurified PCR products did not yield any colonies when transformed. |
| | | | |
| - | There were many colonies on the 6I ligation heat shock (but not electroshock) plate. However, there were of insufficient size to work with, so they were left to grow for another day (or 2, considering how the power will be out on the 18th).
| + | The plans for the metA construct induction were further discussed. Overnight colonies will be made of the construct and a negative control, then they will be diluted in 1 in 100 LB, allowed to grow to 0.4 OD, induced with 0.1 mM new IPTG, and harvested at a certain OD following this, then run on an SDS PAGE. This will test to see if the IPTG really would stimulate the production of the metA gene. The negative controls were not in place yet. Four potential constructs have been grown up overnight, and are labeled 1-4 on tape in the right section of the left shaker. |
| | | | |
| - | The plans for the metA construct induction were further discussed. Overnight colonies will be made of the construct and a negative control, then they will be diluted in 1 in 100 LB, allowed to grow to 0.4 OD, induced with 0.1 mM new IPTG, and harvested at a certain OD following this, then run on an SDS PAGE. This will test to see if the IPTG really would stimulate the production of the metA gene. The negative controls were not in place yet. Four potential constructs have been grown up overnight, and are labeled 1-4 on tape in the right section of the left shaker. Is there some label on the metA protein that we could use to do a Western?
| + | The data from the supernatant growth experiment was partially analyzed. It appears that 14 hours was not sufficient to reach a final OD in some cases, but there was growth in many of the wells. It was also suggested that we could just test the relevant amino acid concentration using HPLC. |
| - | | + | |
| - | The data from the supernatant growth experiment was partially analyzed. It appears that 14 hours was not sufficient to reach a final OD in some cases, but there was growth in many of the wells. It was also suggested that we could just test the relevant amino acid concentration using HPLC. One thing that I was wary about concerning this is that the HPLC would only pick up pure amino acid, and if there was, for instance, a short protein fragment, it could be used for growth, but not show up on the HPLC. Any thoughts? | + | |
| - | | + | |
| - | The cultures for the MinteinC (As it shall henceforth be known) grew well, but were not miniprepped. Once they are miniprepped, then site directed mutagenesis must be done. The cultures of the potential yddg biobricks grew well, and will be confirmed either by PCR of the plasmid using VR and VF2, or by restriction digest.
| + | |
| | | | |
| | From our previous experiments concerning the consortia syntrophy, we saw that three cultures inoculated straight from LB into minimal M9 led to a mixed culture OD that reached about 1.3, similar to 0.1 mM (saturating) Met for metA-, and 0.1 mM Trp for TrpA-. We suspect that this is due to syntrophy, but have yet to do the relevant tests to see if 1 in 100 LB actually does supply significant amounts of amino acids. | | From our previous experiments concerning the consortia syntrophy, we saw that three cultures inoculated straight from LB into minimal M9 led to a mixed culture OD that reached about 1.3, similar to 0.1 mM (saturating) Met for metA-, and 0.1 mM Trp for TrpA-. We suspect that this is due to syntrophy, but have yet to do the relevant tests to see if 1 in 100 LB actually does supply significant amounts of amino acids. |
| - |
| |
| - | I will be going on vacation for the next couple of weeks, and will return in September.
| |
| | | | |
| | - [[User:jacobtoth|Jacob Toth]] | | - [[User:jacobtoth|Jacob Toth]] |
| | | | |
| - | | + | <div style="text-align: center;"><h2>[https://2012.igem.org/Team:British_Columbia/ConsortiaTunability Back to top]</h2></div> |
| - | | + | |
| - | [[File:Joe1Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | [[File:Joe2Team_british_columbia_2012.png|centre|400x500px]] | + | |
| - | | + | |
| - | [[File:Jacob1Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:Jacob2Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:Jacob3Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:Ruichen_Xue_1_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:Ruichen_Xue_2_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:Mehul1_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | [[File:Joe_4degroom_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:4degroom_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | [[File:Plate_reader_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | [[File:Hallam_Lab_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | [[File:ConsoriaCells_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:ConsoriaCells2_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | | + | |
| - | [[File:ConsoriaPlates1_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:ConsoriaPlates2_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:ConsoriaPlates3_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | [[File:ConsoriaPlates4_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | | + | |
| - | [[File:ConsumptionRateMeasurement_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | | + | |
| - | [[File:RunningHugeGel_Team_british_columbia_2012.png|centre|400x500px]]
| + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | | + | |
| - | - [[User:Ruichen|Ruichen]]
| + | |
July 15
An attempt at doing an experiment to calibrate the fluorescent proteins for the plate reader (looking for quenching, linear relationship between OD and fluorescence, etc) was done on Sunday, but as it turns out, we had the wrong promoter for a fluorescent protein, and the wrong antibiotic resistance for one of the other proteins. Given this, we gave up, and decided to do it later with the proper strains.
Several new combinations of fluorescent genes, antibiotic resistances, and Keio strains were made. We used the wrong ones last weekend, but now, we are going to test TyrA- +GFP, TrpA- +RFP, MetA- +YFP, and also wildtype with the fluorescent genes. Recently, we switched backbones for the 13M RFP to Psb1C3, for we do not know the provenance of our current psb1C3 with rfp (we suspect it might be on the lac operon, as per the registry guidelines), and we also put the GFP on the psb1C3 instead of a kanamycin resistant plasmid, because we were transforming into kanamycin resistant auxotrophs.
Just for qualitative fun, we are trying to grow the ArgC- and the ArgE- together in M9 to see if anything results, with negative controls for both. We also tried growing a wildtype with a TrpB auxotroph with rfp to look for qualitative red fluorescence. The fluorescence calibration experiment must be done before we can do these sorts of things quantitatively.
- Jacob Toth
July 19
We tested the growth rate of our 3 main auxotrophs in various (log scale from 100 pM to 10 mM) concentrations of the amino acids to do monod kinetics, which will allow us to predict growth rates at various concentrations of the amino acids.
- Jacob Toth
July 20
Obtained the data from the 3 main auxotrophs growth rate dependences of amino acid concentration.
Obtained the reading of fluorescence-population correlation plate.
- Ruichen
July 22
Joe did 5 more fluorescence-population correlation plate, though found out that the yfp is not expressing correctly.
- Ruichen
July 23
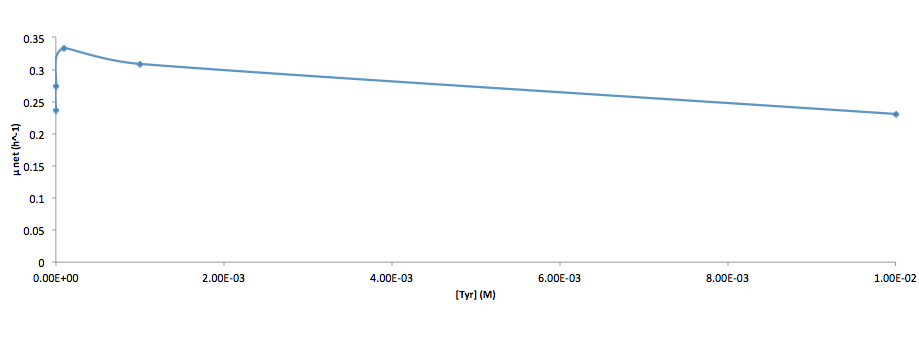
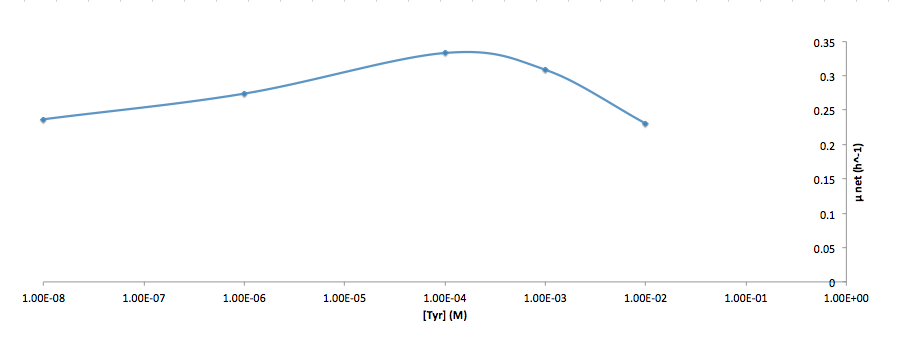
Analyzed the data, and found out that the growth rate and the amino acid (Tyr) concentration has a correlation as follows:
- Figure 4. cell growth-rate at different Tyr concentrations
- Figure 5. Log scale of cell growth-rate at different Tyr concentrations
It was found that the cells depleted the nutrient quickly at low Tyr concentration, and for future experiments it is suggested to use lower initial cell concentrations to extend the time of its exponential growth phase. Also, the wild type has almost the exact growth rate as the autotroph when the the Try concentration is exceptionally high (0.01 M).
- Ruichen
July 26
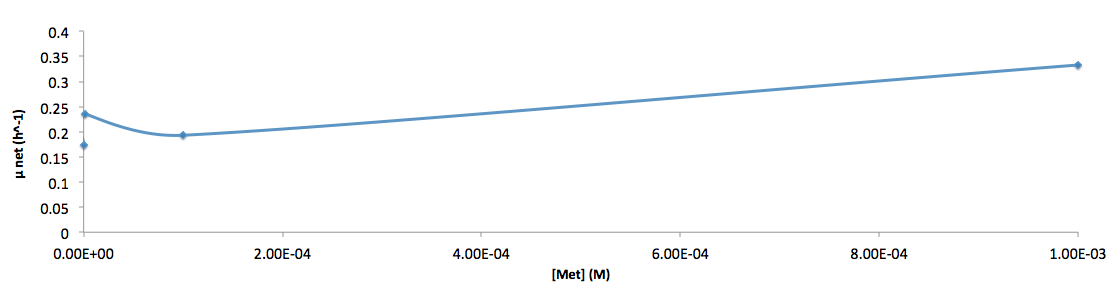
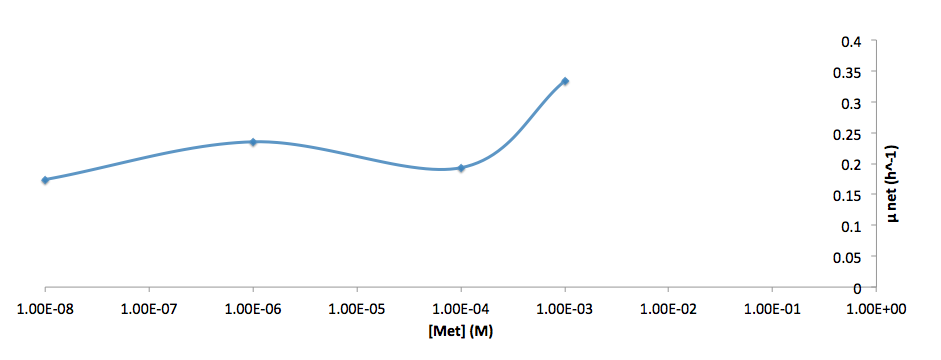
The growth rate and the amino acids (Met and Trp) concentration has a correlation as follows:
- Figure 6. cell growth-rate at different Met concentrations
- Figure 7. Log scale of cell growth-rate at different Met concentrations
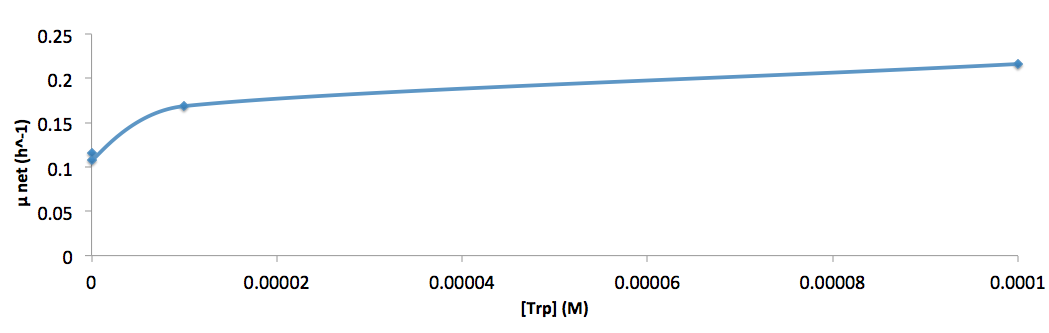
- Figure 8. cell growth-rate at different Trp concentrations
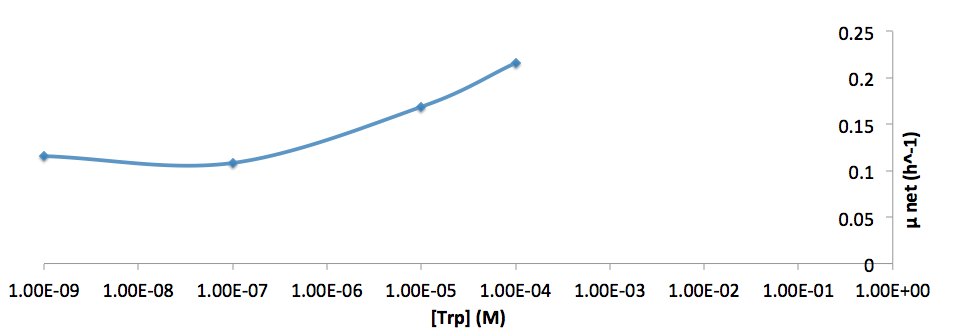
- Figure 9. Log scale of cell growth-rate at different Trp concentrations
- Ruichen
August 3rd
Started to work on the data obtained on July 28th plate readings.
Eventually arrived at the following graphs of growth rates observations, when n=3
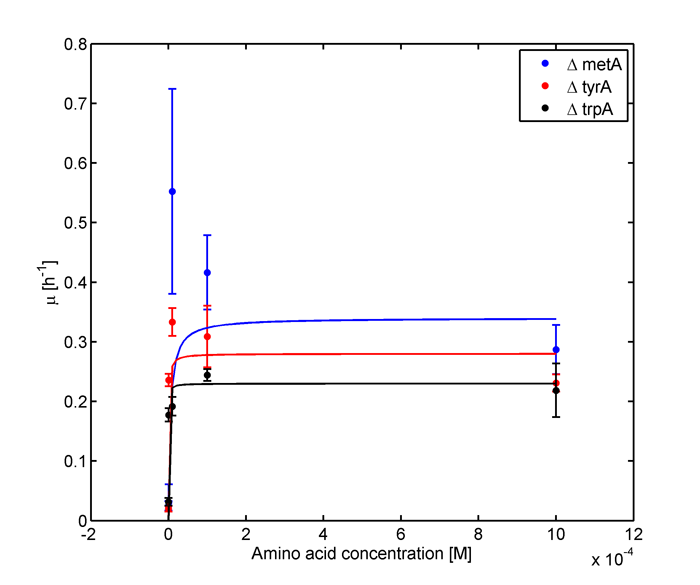
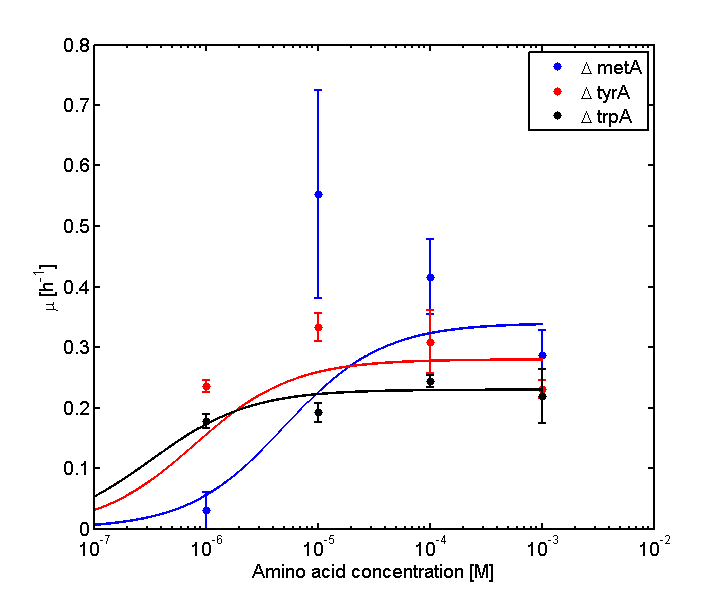
From these graphs, the maximum growth rate, and Ks values that are used in Monod Kinetics can be determined.
- Ruichen
August 12
With a set of the primers that arrived, we were able to PCR amplify certain parts that will be able to be Gibson assembled. On the plasmid that has RFP (a modified BBa_K093012, placed on the Psb1C3), we wanted to put arabinose inducible MetA or arabinose inducible TyrA. However, one of the primers necessary to amplify the plasmid itself, which would be necessary to incorporate the proper homologous sequence for Gibson assembly, did not arrive. Additionally, the PCR to amplify the MetA gene did not work the first time, but has been repeated. The arabinose promoter was successfully PCR'd out of the E. coli genome for both cases.
For the plasmid with YFP, the BBa_I13973, we wanted to put rhamnose inducible TrpA or TyrA after the YFP gene. We received all the primers necessary for this, but the PCR for the plasmid backbone gave several bands and was repeated at a higher annealing temperature. The results are not yet known. The rhamnose promoter was successfully PCR'd out of the genome, as was the TrpA gene. However, we lacked one of the promoters for the TyrA gene. Since there were several bands for the YFP plasmid, we did not do a Gibson assembly. Additionally, it is possible that the primers were designed for the pSB1C3 rather than the pSB1A2, but that should have little effect due to the similarities on the ends of the two plasmids. A simple digest and ligation should be able to move the part from the one plasmid to the other.
We also wanted to put a IPTG inducible (BBa_K091111, LacIQ) metA gene after a GFP. The PCR for the MetA gene and the GFP plasmid were successful (although the GFP plasmid appeared as a fairly weak band on the gel), but the PCR for the LacIQ promoter failed. The initial DNA for this PCR was added directly from the parts distribution kit, and as such the composition and concentration of exactly what was added was unknown. The PCR was repeated as a cPCR on the genome, so as to at least get a usable part with the right overhang.
In an effort to see how much amino acid each type of cell in the co-culture will produce, we have been harvesting supernatants of our normal cultures as per the protocol in Kerner et al. Once we have supernatants of the cultures at various ODs, we are going to grow the auxotrophs in this supernatant, and then look at the growth rate and final OD.
Joe has found inconsistent results with his fluorescent proteins on the various promoters, and we have plans to standardize the promoter and rbs for the fluorescent proteins that will be used. Additionally, there is some overlap between YFP and GFP that is proving difficult to account for, so we are going to try to switch one to either BFP or CFP.
We also wanted to grow three of the consortia together to see what sort of final OD they reach, and have been monitoring it sporadically for the past 24 hours.
- Jacob Toth
August 13
Having harvested the supernatant of the various cultures at various ODs, we filter sterilized it with a 0.2 uM filter to remove any cells that may have still remained. Now that we have the sterilized supernatant, we are going to follow the protocol in Kerner et al 2012, and add 1/5 5X M9 media, and grow different auxotrophic cultures, noting the OD and growth rate. From this, we will try to calculate, or at least approximate, the amino acid export rate of the cells.
The data that we collected regarding the growth curves of the auxotrophs and the mixture are very different from what the plate reader previously showed. We think this may be because of a lack of correction for path length by the plate reader, but aren't entirely sure.
- Jacob Toth
August 15
After two days of shaking at 37° in 1 mM IPTG, the supposed IPTG inducible metA constructs in the metA auxotrophs were unable to grow.
- Jacob Toth
August 16
The supposed metA constructs still appears unable to complement a metA knockout. It has been suggested that the concentration of metA may have been too high, and this would have led to a severe detriment to the cell, and it has also been suggested that the IPTG, being a rather old stock, was no longer good. Another step that we are going to take is to see if the expression of the construct can be seen on an sds page under induction.
Concnerning the other constructs, the PCR for the YFP has yet to give us a single solid band, so we are trying to move it into the psb1c3 plasmid in case there are some plasmid specific sequences that were giving us poor results. The ligation and transformation has been done, and we are waiting for colonies. The PCR products necessary for the gibson assembly of the GFP-IPTG-TrpA gene were also all available, and that gibson assembly has been done. However, due to the failure of several gels, there was very little product left, and PCR purification would likely result in unusuable quantities, so we tried a Gibson assembly with the straight PCR product.
Another PCR was done to generate some of the other parts required for assembly of other parts. The PCR for the arabinose promoter (using a new primer) has not yet resulted in a band.
There are two distinct bands on the RFP PCR, one at the correct length, the other much lower. Several other products were also generated today, but there are still two essential parts, that of YFP and that of the new arabinose promoter, which have not yet been successfully PCR'd.
- Jacob Toth
August 17
The Gibson assembled parts using unpurified PCR products did not yield any colonies when transformed.
The plans for the metA construct induction were further discussed. Overnight colonies will be made of the construct and a negative control, then they will be diluted in 1 in 100 LB, allowed to grow to 0.4 OD, induced with 0.1 mM new IPTG, and harvested at a certain OD following this, then run on an SDS PAGE. This will test to see if the IPTG really would stimulate the production of the metA gene. The negative controls were not in place yet. Four potential constructs have been grown up overnight, and are labeled 1-4 on tape in the right section of the left shaker.
The data from the supernatant growth experiment was partially analyzed. It appears that 14 hours was not sufficient to reach a final OD in some cases, but there was growth in many of the wells. It was also suggested that we could just test the relevant amino acid concentration using HPLC.
From our previous experiments concerning the consortia syntrophy, we saw that three cultures inoculated straight from LB into minimal M9 led to a mixed culture OD that reached about 1.3, similar to 0.1 mM (saturating) Met for metA-, and 0.1 mM Trp for TrpA-. We suspect that this is due to syntrophy, but have yet to do the relevant tests to see if 1 in 100 LB actually does supply significant amounts of amino acids.
- Jacob Toth
 "
"










