 "
"
Team:Waterloo
From 2012.igem.org
In Vivo Protein Fusion Assembly Using Self Excising Ribozymes
ABSTRACT
Waterloo's 2012 iGEM project is a continuation of the 2011 project, In Vivo Protein Fusion Assembly Using Self Excising Ribozymes. This year our hope is to complete the project with the aim of potentially designing future projects which incorporate this system.
Self-excising ribozymes are RNA sequences with catalytic properties which allow them to remove themselves and the regions which they flank from an RNA sequence. These are introns; however, with ribozyme self-excision the introns are removed without the aid of protein enzymes. In our project we use self-excising ribozymes to remove an extraneous sequence, an intron, which interrupts the coding sequence of GFP. Upon successful removal of the intron, the two halves of GFP should be ligated together and be able to be translated into a fully functional GFP. By showing that functional fusion proteins can be assembled in-vivo using this system we open up possibilities such as the addition of recombination sites to allow gene shuffling, and regulatory sequences which function at the DNA level but that are removed at the RNA level to create functional proteins.
WE WOULD LIKE TO THANK OUR GENEROUS SPONSORS.

with Mathematical Modelling
1.0 Abstract
The Group 1 introns are a widely spread group of ribozymes. Many of them exhibit self-excising activity. These could be used to generate in vivo recombination libraries by incorporating them with a Cre-lox system, which can facilitate chromosomal gene transposition. Below we outline a proof-of-concept experiment to display the feasibility of this experiment.
2.0 Introduction
2.1 Cre-lox system
The Cre-lox system is an in-vivo recombination system developed from P1 bacteriophage that is perhaps most familiar to students as a method of gene self-excision (1). The Cre enzyme is a site specific recombinase. It recognises loxP sites in DNA (34bp), and will either excise, invert or translocate DNA between them (Fig 1). The Cre enzyme may be designed for expression under specific conditions (eg. IPTG induction), allowing some control over in vivo recombination. The caveat to using any recombination system to generate fusion proteins in vivo, is that translation of the leftover recombination site sequence or 'scar' can shift codon readings, and/or disrupt protein folding (Fig 2) We propose, therefore, to flank a recombination site, such as the loxP sequence, with a self-excising ribozyme sequence, to create 'scarless' fusion proteins (Fig 2).


Fig 2. Schematic of recombination products a) with a remaining 'scar' and b) with the scar removed from between two fusion protein segments, PL and PR. Failure to remove the lox scar may result in disrupted protein folding or mistranslation of the second half of the protein, PR
2.2 Group I Introns
Self-excising ribozymes are an ancient class of introns that can remove themselves from RNA constructs, including mRNA and tRNA (3). The Group I introns are a subset of these self-excising ribozymes, and are widely distributed through simple eukaryotes, fungi, mitochondria, chloroplasts, bacteria and bacteriophage (3). Group I introns are not yet known to provide a specific biological function, save to excise themselves from important RNA sequences, and thus prevent host death. One hypothesis is that they are "selfish" remnants of an RNA world (4). Some Group I introns, for example, code for DNA endonucleases that help them migrate to new sites (3).
The Group I excision mechanism relies on a reactive 'core' rich in RNA secondary structure that conducts two successive transesterifications (5). Some introns require proteins to help stabilise them, while others are completely independent (5). The first transesterification involves a nucleophilic attack on the 5' end of the intron, by the 3' OH of a guanosine nucleotide (5) (Fig 3). Structural rearrangements bring the the 3' end of the exon into proximity with the 3' end of the intron, allowing the second transesterification to occur (5) (Fig 3). The intron is usually then degraded by the host, while the resulting mRNA is translated.

Fig 3. Mechanism of intron self-excision (adapted from (5)) – The construct used above is our experimental construct, further discussed below. (a) The first transesterification occurs when a GMP attacks the 5' end of the intron (InL-lox-InR). (b) The second transesterification occurs at the 3' end of the intron, led by the 3' end of the exon. This results resulting in (a) the fusion mRNA for transcription and (d) the intron sequence, to be degraded.
2.3 Staphylococcus phage twort ORF142
The Staphylococcus aureus bacteriophage Twort is notable for having three introns within a single gene (6). This ORF142 is a putative structural protein, though its role has not yet been confirmed. Sequence analysis reveals three highly similar introns, I1, I2 and I3, closely interspersed in the gene. Experiments show that they are self-excising, and can produce variable splice products, depending on the intron(s) removed (6). This is likely due to their terminal sequence similarity (6). We have chosen to work with a modified version of these introns due to their independent, self-excision capability, and the low likelihood of disrupting important secondary structure on incorporation of additional RNA.
2.4 Application
Gene shuffling has been used to create fusion protein libraries of compounds that are more effective than the parent proteins. Examples include interferon, antibodies and Cry proteins, a family of biological insecticides (7). Many of these recombination systems, however, rely on in vitro recombination, cloning, then expression and screening of the protein products. An in vivo recombination system allows the recombination and expression to happen in the same step.
3.0 Lab and Design
Our project, which continues from last year, is a proof-of-concept for the use of Group I introns in providing scarless removal of an internal sequence. The marker gene we are using is GFP. Fig 3 (above) and Fig 7 (below) describe the desired, final construct. Essentially, successful self-excision of the intron should result in a normal, functional GFP protein. The final construct will be assembled by joining five "pieces": the two halves of the GFP protein, the two halves of the intron, and a lox recombination site.
3.1 Mathematical model
In order to characterize the relative efficiency of the intron system, a model was developed based on the idea proposed by Kelly et al (8) of comparing steady state fluorescence to a common standard. Our model develops a ratio of protein expression in a system with the intron (Pintron) (Fig 4), to expression in a system without the intron (PGFP) (Fig 5). It assumes that all reaction rates are linear, and that they remain constant between cell populations. If the rate of mRNA degradation (δM) is known or estimated, one can approximate the rates of successful mRNA splicing (αS) and failed splicing or folding (αF). The resulting measure of efficiency of splicing, then, is αS / (αS + αF)

Fig 4. Model predicting amount of protein formed from a construct containing an intron

Fig 5. Model predicting amount of protein formed from an intron-less construct

Fig 6. The ratio of the rate of protein synthesis with and without an intron
3.2 Procedure outline
We have designed the construct to be built using the meta format of RFC53 in pSB1C3 and pSB1C5. RFC53 is useful for creating scarless recombination between DNA sites using the REN EarI. This cleaves a set distance downstream of their recognition site.
First, the pieces must be subcloned from pUC57 into pSB1C3 or pSB1C5, which contain the meta-suffix format of RFC53. The planned construct maps are shown in Fig 7.

Fig 7. Planned constructs (a) main experimental construct (K576011), (b) positive control (K576013), (c) negative control (K576012) in pSB1C3
Last year, a diagnostic of the final ligation products revealed unexpected banding. This year, we have re-done the subcloning into pSB1C3, and have proceeded to attempt the ligation of the final construct with more caution. Previously, diagnostic gels were run only insofar as gel extraction had to be done. As such there was no confirmation of the ligation pieces before joining them. This was in part due to the size of the fragments, as several of them are quite small, making it difficult sometimes to visualize them on a gel.
3.3 Lab work, results and conclusions
This year, in addition to conduct the expected laboratory research on the intron project, we invited 20 undergraduate students from the University of Waterloo to join us, in order to teach them some basic molecular biology techniques. Volunteers learned simple molecular biology techniques such as restriction enzyme mapping, miniprep plasmid samples, vector cloning, heat-shock transformation and agarose gel extraction.
The original parts received by our team include left intron (aka Intron 1), right intron (Intron 2), left GFP (GFP 1), right GFP (GFP 2) and a lox region of 34 bp in length. All parts were cloned into pUC57 vector backbone, which is ampicillin resistant. In order to conduct the RFC 53 protocol in our cloning procedure, we need to first sub-clone the R-Intron, L-Intron, L-GFP and R-GFP into pSB1C3 backbone (Chloramphenicol resistant or CmR).
To complete the sub-cloning process, we followed a flow-chart as shown in Figure 8(a)

We used REN EcoR1 and Pst1 to double digest the original parts and then cloned into pSB1C3 vector. We transformed the cloned vector + insert samples into competent DH5α strain of Escherichia coli and then plated the colonies onto plates with Chloramphenicol.
Controls are important for plasmid cloning; in addition to our transformation plates, we included vector+ligase control, vector-only control, transformation control and negative (no plasmid) control to check for possibility of contamination and improper techniques. As expected, the transformation control, vector+ligase and vector-only controls had only pink colonies growing on them. The negative control plates were clean.
Our transformation plates showed a mix of pink and white colonies; the pink colonies represent the presence of Red fluorescent Protein in pSB1C3. As the multiple cloning site for pSB1C3 lies inside a red fluorescent protein (RFP) gene, successful transformants are white due to disruption of the RFP. We screened 8 of these colonies (2 colonies for each type of insert) and checked the size of the inserts by running an agarose gel, as shown in Figure 8 (b). In addition, we also loaded the digested pUC57 samples to ensure the inserts look identical. The ladder we used in this agarose gel is Fermentas GeneRuler 1kb DNA ladder. We have saved 2 copies of each of 8 clones in our lab freezer and we have also sent one of each type of clone (a total of 4 clones) to the IGEM registry.

After successfully cloning the inserts into pSB1C3, we proceed with the RFC 53 protocol to make the main construct. Unfortunately, we were unable to find a clone with the right-sized insert for the L-intron and L-GFP ligation due to time constraints. As a result, we were unable to finish the making of the main construct.
The positive control construct is assembled using RFC 53. Fluorescence is expected to demonstrate that the one amino acid scar from RFC 53 does not disrupt GFP. The negative control construct is assembled using RFC 10. Fluorescence is not expected, in order to demonstrate that presence of the lox site disrupts GFP folding and activity.
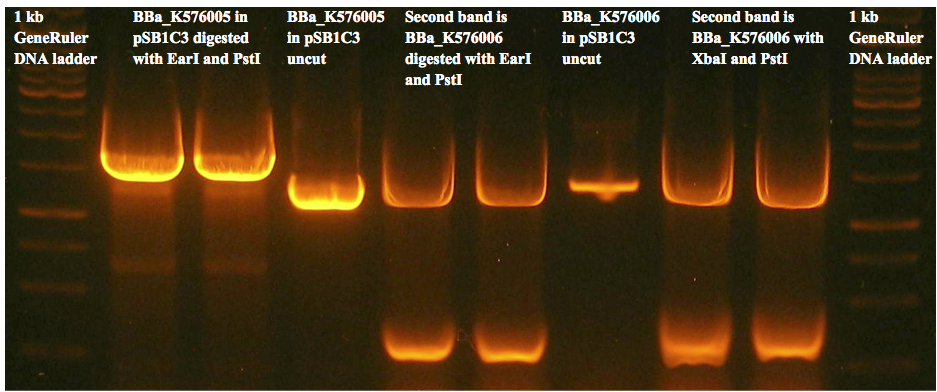
Figure 8(c) is an example of a gel used to prepare gel extracted fragments for the two control constructs. We were unable to get any successfully transformed colonies for either construct.
Two criticisms in last year's experimental design have been highlighted this year, which may explain some of the difficulties in creating a working construct. First, many of the fragments being used are relatively small, which makes it difficult to isolate them for ligation. A suggested solution is to use PCR to amplify or introduce, for example, the 34bp lox site to the construct. Second, some of the REN being used for double digests are very close to each other on the plasmid (< 10bp). This may be impeding the RENs' ability to bind and cleave DNA. An alternative design to the one proposed here would be to create a gene with multiple intron sites to allow continuous mRNA recombination in vivo, rather than a one time recombination event at the DNA level (6).
3.4 Side project: Promoter expression quantification
In parallel with the lab work of constructing the intron system, progress was made at our lab towards implementing a framework for measuring relative fluorescence of cell cultures, using the protocol described by Kelly et al. (8). Such measurements are necessary in order to quantify the notion of intron efficiency developed in our model.
Work on this project is ongoing. The steps taken this summer were:
• identification and acquisition of appropriate materials and equipment access
• training of four undergraduate students, separate from the main lab project, in essential lab techniques
• test-running the protocol and troubleshooting errors.
Once a quantification system is successfully implemented, our team will be also be equipped to begin making measurements of many other registry parts, according to the RPU standard proposed by Kelly et al. (8). Our vision is for RPU quantification of existing registry parts to be a standard component of UW iGEM's yearly activities, beginning with constitutive promoters and expanding into measurements of more complex devices.
References
(1) Nagy, A. (2000). Cre Recombinase: The Universal Reagent for Genome Tailoring. Genesis, 26(2), 99–109.
(2) The Jackson Laboratory. (n.d.). Introduction to Cre-lox technology. (http://cre.jax.org/introduction.html)
(3) Haugen, P., Simon, D. M., & Bhattacharya, D. (2005). The natural history of group I introns. Trends in Genetics, 21(2), 111–119.
(4) Lambowitz, A. M., Caprara, M. G., Zimmerly, S., & Perlman, P. S. (1999). The RNA World, Second Edition. New York: Cold Spring Harbor Laboratory Press, 451–486.
(5) Cech, T. R. (1990). Self-Splicing of Group I Introns. Annual Reviews in Biochemistry, 59, 543 – 568.
(6) Landthaler, M., & Shub, D. A. (1999). Unexpected abundance of self-splicing introns in the genome of bacteriophage Twort: introns in multiple genes, a single gene with three introns, and exon skipping by group I ribozymes. Proceedings of the National Academy of Sciences of the United States of America, 96(12), 7005–7010.
(7) Glick, B. R., Pasternak, J. J., & Patten, C. L. (2010). Molecular Biotechnology: Principles and Applications of Recombinant DNA, Fourth Edition. Washington DC: ASM Press.
(8) Kelly, J. R., Rubin, A. J., Davis, J. H., Ajo-Franklin, C. M., Cumbers, J., Czar, M. J., de Mora, K., et al. (2009). Measuring the activity of BioBrick promoters using an in vivo reference standard. Journal of Biological Engineering, 3:4.
UW iGEM OUTREACH PROJECTS 2011-12
Our Mission: Raise awareness on issues concerning synthetic biology to make informed, fact-based opinions.
Introduction
This year’s UW iGEM’s Outreach sub-team has learnt a lot from its experiences. We were met with a few hurdles with regards to planning and implementing the myriad of events we had hoped to have to engage our community. Though with every step there was much to be learnt and we have decided to discuss all the great events we did have planned and are in the process of planning. Along with that we thought it would be a great resource to create a guide that discussed various different types of outreach events you can hold and what hurdles you could potentially face (as we did) in terms of implementation.
Successes Despite the barriers that we faced, we still had numerous successes. We were still able to develop Community Bricks for most of our programs so other Outreach members can implement them as well. The Community Bricks we were able to post include:
- 1. Grade 11 Synthetic Biology Workshop Program with Interactive Activities
- 2. DIY, At Home Gel Electrophoresis
- 3. Basic Lab Training Workshops
- 4. Communications Plan Template and Example for Your Large-Scale Outreach Programs
- 5. Tips and Tricks to Have a Successful Outreach Event
As well as…
Educational Video Podcast featuring the iGEM Outreach Lead Ekta Bibra on Synthetic Biology for Virtual Researcher On Call (organization providing educational resources to students and teachers through online web conferences or podcasts in Ontario, Canada). This material will be openly available for students and teachers to view and learn from across the province. This will be available for you to view at the regionals this year!
As well, we are working on a core social media campaign in which we amplify awareness with minimal costs, gain a larger audience base to ease overall promotion and easily apply Outreach’s over-arching goal of raising awareness on synthetic biology. Make sure you follow us and our progressions on:
Twitter: @Waterloo_iGEM
Facebook: Waterloo iGEM page
Outreach Activities
The divisions of our outreach events are regularly divided into seasonal timings, from Fall, Winter to Spring. This year these were the events that we had initially planned on implementing:
- BioTalks Open Panel Discussion and Lecture Series Community Brick (Communications Plan Plus Example)
- Engineering Science Quest (ESQ) View Community Brick
- Hot Chocolate Event Purpose: To spread the word on campus of UWaterloo iGEM, gain presence amongst students of who we are and promote our social media accounts on Twitter and FaceBook.
- Movie Night Purpose: In a social setting, show what types of content regarding biotechnology and social media are presented to the public. Demonstrate how that affects the overall perspective that they have towards the topic and how it shapes their future opinions.
- Basic Lab Training
Community Brick
Purpose: Since we are a co-operative education school (meaning many students within our University do 5 or more 4 month internships with employers) it helps to have a kick-start on basic lab skills and techniques to add onto their resumes when they may not have that experience yet. This will help to make them more competitive when applying for positions.
How: Hold a one day training session where University students are taught basic skills used everyday in our wet lab.
Type: Education - Grade 11 Synthetic Biology Workshop
Community Brick
Purpose : Similar in nature to UW iGEM 2011 Outreach’s Grade 12 Synthetic Biology Workshop (you can find the Community Brick for this, this interactive workshop’s aim was to promote open discussion on industries and current issues within synthetic biology.
How: Hold a workshop using lab space and the development of two new activities that would suit the Grade 11 Biology curriculum.
Type: Education - Tips and Tricks to Have a Successful Outreach Event
Community Brick
Purpose : To take what has been learnt throughout the year and streamline the key values into a valuable guide for those interested in pursuing outreach activities.
How: Develop a brief information sheet that puts together key points for organizing outreach events.
Type: Educational
Purpose: Develop an exciting discussion forum between science undergraduates and industry specialists within biotechnology to encourage science entrepreneurship, innovation and industrial advice.
How: Organize a large-scale open panel discussion involving brief 20 minute talks from key industry leaders followed by a question and answer period that can supplement what has been demonstrated within the short lectures.
Type: Education/Awareness
Purpose: A tradition for the UW iGEM Outreach team to carry out, this workshop is held by ESQ to develop hands-on learning activities and workshops for students from Grade 3-12 to promote science/engineering education and outreach.
How: Development of a do-it-yourself workshop for Grade 12 students that incorporates science into their everyday lives. Also created a Community Brick.
Type: Education
How: Selling hot chocolate for 50 cents along with a small paper tag of our FaceBook and Twitter page.
Type: Marketing/Promotion
How: Show a very popular movie that has recently played or a well-known classic to the students at the University.
Type: Awareness
IN DEPTH DESCRIPTION OF EVENTS:
- BIOTALKS OPEN PANEL DISCUSSION
Community Brick (Communications Plan Plus Example)
This event we are planning will be more than just a lecture series, but it is meant to be so much more. It is an experience, a place of discussion and intellectual exchange and of learning. Those are some of the visions we have for this event.
By having key industry speakers do brief 20 minute lectures on topics such as: Clinical Trial Drug Development, Commercialization of Biomass and Energy Products and Entrepreneurial Barriers for Biotechnological Companies, we are hoping to bridge the level of discomfort scientists have when it comes to business. We want to encourage innovation amongst the highly technical individuals within our university and provide an open forum of educational resources for them to gain knowledge.
Currently, UW iGEM has a core team of members working on this initiative and we are hoping to implement the BioTalks event in March 2013. We have completed our communications plan, gathered 3 speakers from the Pharmaceutical, Agricultural and Renewable Energy sectors and have separated the organizational team into three components: Director and Lead of Social Media Marketing (Ekta Bibra), Internal Communications and Logistics Lead (Anjali Arya) and Vision and Strategy Lead (Angela Biskupovic). Now we are working on meeting the appropriate potential sponsors and advisors.
The creation of a communications plan is very helpful in creating clear-cut objectives and values, giving your team an over-arching mission and have stakeholders who you are proposing this event to understand exactly what you are trying to do. We have created a Community Brick that demonstrates a template for writing a Communications Plan as well as our BioTalks example that we are currently proposing to potential sponsors and advisors.
Our next steps involve, the development of an aggressive marketing strategy through social media and direct marketing, raising funds and all the logistical work involved with a large-scale event such as this. Please stay tuned! If you have questions on this event feel free to contact us and we would love to share ideas with you!
Contact the BioTalks team at: uwigem.outreach.hp@gmail.com (Ekta Bibra, Outreach Lead) - ENGINEERING SCIENCE QUEST (ESQ)
- HOT CHOCOLATE EVENT
- When serving food publically, one must first contact Feds and let them be aware of your ideas to host such event well in advance (estimated 4 weeks prior). Space is very limited on campus therefore letting Feds know of your plans somewhat secures your spot on campus for that day and time.
- Next comes having the Waterloo Health Region approve your plans and your food before your serve them to the public. This is a fairly long process and usually teams/clubs/groups are advised to fill out this form and get in touch with Waterloo Health Region at least 8 weeks prior to your event.
- While you are still waiting to hear from the Waterloo Health Region, make sure you keep in touch with what else is going on around campus that week that could possibly interfere with your event. For example I our case, UW’s Welcome Week BBQ lunch was being held at the same time same area, same day. Try and contact those individuals so you can work out the space and equipment (if sharing) so no issues arise on the day off the event.
- In addition, have your ingredients (same ones listed on the WHN form) arranged and readily available on the day of your event.
- Lastly, to save yourself time, if you will be using electronic equipments or large equipments such as water thermals that require you to bring hot water externally, have it arranged close to your location and already approved by the “company” supplying it to you.
- Throughout this whole process keep in mind that communication is key. You will be in touch with various individuals from various groups and so be as descriptive and detailed as possible to get your point across.
- MOVIE NIGHT/ENTERTAINMENT NIGHT
- LABORATORY EVENT Community Brick Available for Protocol
- EXTERNAL WORKSHOPS
Outreach took a part in the Engineering Science Quest at University of Waterloo by holding a lab activity for high school students (ages 14-17). The idea was to show students that it is easy to do Science at home.
Gel Electrophoresis was performed using all supplies available at home (except for agarose- but one can use Jello too). We performed this activity with 3 different groups of ~10 students each time over the span of 2 months. We paired students together and gave them a chance to work in a team and learn some basic lab techniques such as pipetting, understanding the idea of having a medium for the electrons to flow in and much more.
Students were also given a formal presentation at the end of the activity where the theory of Gel Electrophoresis was explained and were at the end asked a few questions for small prizes.
Students were able to separate food dye (based on their charge) successfully; as 4 out of 5 groups, on average, did see a proper separation of colours.
The purpose of this event was to bring the UW campus community together and educate them about the field of synthetic biology through iGEM.
The idea was to serve hot chocolate to individuals wishing to buy it for 50 cents a cup. While they are getting their drink served to them, iGEM members will be there engaging in conversations with the various students and faculty, discussing with them the history of synthetic biology and how it has evolved over the years.
Hosting such an event especially a one that involves serving food to the local public, requires a lot of permission seeking for various personnels.
The following protocol must be followed at UW in order to serve food publically anywhere on campus:
The purpose of this event was to show a movie related to Synthetic Biology in relation to what is new today (for a possible discussion later on).
When starting an outreach event involving entertainment for the first time one must. Understand that the turnout may not be very high but remember it is a first-time event. Especially if it is held during midterm period.
Next you must focus on getting in touch with Feds again and communicating with them to try and find a place where not only can you show a movie but also accommodate for the amount of people you expect to turn up for this event. This can be tricky if you have a very specific date picked out. Always have a range of dates as a backup plan because since space if limited on campus, you might not always get your desired location at the day and time you want it.
If there are specific individuals in-charge of these events make sure they are aware of not only where to find the appropriate equipment needed to show the movie on campus but also the process involved in the rental. There are very strict protocols that need to be followed in regards to electrical equipment being rented at UW. Get in touch with media doc. for more details for UW.
Another thing to keep in mind is that University of Waterloo is only allowed to show licensed movies/videos on campus. Feds is usually a great help in this process. They tend to have a huge list of movies available for you to choose from and in most cases the movie you want to show will be included in that licensed list. If not try and get in touch with one of the Feds executives or clubs manager to see if they can assist you in getting a license to show that movie on campus. The movie however will be rented from the closest store.
One last thing to be kept in mind is, if your group plans on serving food at this event, make sure to follow the steps similar to the ones listed above for the food event.
The purpose of this event was to educate science students about the basic and complicated lab techniques that are required during their undergraduate years here at Waterloo.
This exercise followed a similar pattern to our ESQ exercises, however are made more directed towards first and second year students.
Since this activity is very hands on, laboratory safety protocol is very important to follow. Each student that wants to participate in this activity must have their WHMIS certification before they show up for the event.
For activities like this only a lab space can be used to perform the experiments. Therefore it is crucial that you get an instructor’s permission to his/her lab space. With regular lab sections utilizing the lab space for a minimum for 3 hours per section, time is very critical. In most cases this even will have to be held when a specific lab is over for the term.
Details of the experiments being performed must be discussed by the instructor before anything is agreed upon. These details should include, the time and date of your event, what kind of experiments the students will be performing as well as what kind of equipment will be provided to them and if necessary what kind of equipment is required by the instructor of the lab to provide for us.
Costs of the equipment and materials being used will also have to be calculated just in case there needs to be a charge placed on this.
Keep in mind that lab spaces are not entirely meant for a large group of students. Prior to deciding on the exact date and time for your event, try and put out a survey to see how many students are actually interested. This will help you decide whether you should have one or two sessions and whether or not they should be on the same time and date or not.
Before finalizing the details for the event just prepare a timeline and run it through the instructor as well as the volunteers teaching the students so everyone is on the same page.
If someone within your team or outside of your team is making a commitment for the event that is critical for its execution, ensure that they either understand that once they make a commitment to a certain time they cannot leave or make them sign a contractual form binding them to the event. This will ensure no last minute cancellations.
The purpose of this event is to education younger generation (Gr. 11 and 12 students) about the field of synthetic biology, what it involves and what kind of future it holds for them. The idea behind it is to let them be aware of the opportunities out there for them in the field of science.
This workshop involves a selected science class from the local high school to come to campus for a whole day event. Being able to contact a High School teacher and letting them know about this opportunity for their students is the main part. It is slightly easier for the beginning year to keep it limited to local High Schools just because that way you are limiting any communication and travelling issues. Team members could also get in touch with their old high school science teacher to see if they are interested to attend.
If the teacher is hearing about this for the first time it is usually beneficial to have brochures, pamphlets or postcards to distribute to the prospects. This makes sure that they leave with enough information to get them to start thinking about the idea of getting their students to come and learn about synthetic biology.
Sometimes getting in touch with teachers can be hard. If you University/College has outreach programs that associate with high schools, get in touch with them, they can be a great aid in terms of networking with the High School teachers.
Keep in mind that even if the High School is local, there needs to be a written contest of the teacher agreeing to perform this activity on the given date and any changes made should be informed to the other party in a well advanced notice. This should also be run through the school’s administration to avoid any last minute changes or cancellations. Set one effective deadline and aim to plan everything accordingly.
In these types of activities communication between you group, the school and the teacher is essential.
After those details have been finalized, similar room booking directions can be followed as listed before.
Motivation and Goals
This year's modelling project focused on extending the work done by the modelling team in 2010.
Waterloo's 2010 iGEM project, "Staphiscope", utilized amplifier parts developed by Cambridge in 2009 to detect low levels of Staph Aureus. These amplifier parts were characterized by the Cambridge team, but only under control of AraC/pBAD promoter, which differed from the promoter used in our 2010 Staphiscope project.
In order to characterize the amplifiers, a parameter scan was undertaken to find promoter-independent Hill parameters of each amplifier, consistent with data of full system. However, empirical verification of our results was lacking. This year, we sought to obtain this data, which (in conjunction with Cambridge data and model), would allow us to find Hill parameters for each amplifier.
Model
To allow for comparison of data, we used the same model as Cambridge in 2009.
In this model, araC represses the pBAD promoter in the absence of the inducer, arabinose. When arabinose is present, it binds to araC, preventing repression of the promoter and allowing transcription of reporter (GFP). This situation is modelled by a Hill function; we seek the Hill parameters of this function.
Thus, when AraC/pBAD system is induced with arabinose, we expect to see a steady increase of fluorescence from a low level, followed by a plateau of fluorescence at steady state.
Method
To measure fluorescence, we closely followed the assay described in the paper "Measuring the activity of BioBrick promoters using an in vivo reference standard", in the section "Assay of Promoter Collections".
Three cultures were grown overnight at 37 degrees Celsius with spinning at 200 rpm: untransformed BW27783, BW27783 containing BBa_I0500, and BW27783 containing BBa_I20260. These were then diluted 1:100 and regrown for roughly 4 hours under the same conditions. They were then diluted to an OD between 0.05 and 0.09, and regrown for 1 hour, again under the same conditions.
After this, the cultures were diluted into a 96-well plate at 8 different concentrations of inducer (arabinose), ranging from 0 to 6.4 uM. The plate was then incubated in a Wallac Victor3 multi-well fluorimeter at 37 degrees Celsius, and repeating measurements of absorbance and fluorescence were taken at 10 minute intervals, with shaking after each measurement. Untransformed BW27783, at each concentration of arabinose, was used to measure background fluorescence, and wells containing only broth were included to measure background absorbance. The machine settings used were identical to those described in the paper referenced above.
With this data, we aimed to calculate the steady-state per-cell GFP concentration during log-phase growth, for both BBa_I0500 and BBa_I20260 (measurement kit for the standard promoter, J23101). The ratio of these values would then characterize the strength of the AraC/pBAD promoter in units of RPU. The justification for this approach can be found in the supplemental material of the paper referenced above.
Results
The results of the experiment were anomalous, and considered too unreliable to be conclusive. There was no clear relationship between cell fluorescence and inducer concentration.
The fluorescence curve did not qualitatively match the predictions of the model; across all concentrations, and for each of the 3 cultures, we observed a high initial fluorescence, with a rapid drop to a lower steady state value. For each culture, this drop in fluorescence aligned well with the growth curve.
In addition, the untransformed BW27783 cells exhibited consistently higher fluorescence than cells containing BBa_I0500, which was highly anomalous. Because of this, we could not reliably use these cells to measure background fluorescence.
Below, a sample graph of Total Fluorescence is shown for each of the 3 cultures. These are curves of the total fluorescence for each culture, averaged over 3 replicates for each culture.

Discussion
It is believed that an error in our strain of BW27783 is most likely responsible for the anomalous qualitative features of our data. This is because for each concentration of inducer, the untransformed BW27783 cells exhibit a fluorescence curve highly similar to that of BW27783 containing BBa_I0500, and yet the untransformed cells should not be expressing GFP.
Prior to the measurement assay, BW27783 cells transformed with BBa_I0500 were plated and examined for fluorescence, both with and without the presence of inducer. The uninduced cells were not found to fluoresce, while the induced cells did fluoresce. The fluorescing cultures were used to make the frozen stock of BBa_I0500 which was used in the measurement assay. This indicates that our untransformed BW27783 should not fluoresce without the presence of inducer. Furthermore, the untransformed BW27783 cells used in the measurement assay were at no point prior to the assay exposed to arabinose.
To explain the fluorescence of the untransformed BW27783 in the measurement assay, it is speculated that our strain of BW27783 exhibits a rapid production of GFP in response to even low concentrations of inducer. Experimental error is also a likely source of inaccuracy in the data, although the qualitative features described were consistent across 3 trials of the experiment. Research into these results is still ongoing.
Determining The Factors Which Affect Perception of Synthetic Biology: A Multiple Regression Analysis
Despite synthetic biology's rapidly growing importance in a wide variety of fields including energy and health, it is still relatively unknown to the population at large. While some may have a vague notion of what synthetic biology is and its potential impact, most do not have anything to associate it with. In fact, some may even find the juxtaposition of artificial (synthetic) and natural (biology) confusing or contradictory. We at the iGEM University of Waterloo Human Practices team believe this represents a prime opportunity to help shape the public perception of synthetic biology and allay the fear and paranoia typically associated with the emergence of similar new fields of study. To do this effectively, we believe it's necessary to examine closely what factors or characteristics may affect a person's perception of synthetic biology. The purpose of this study, then, is to use statistical analysis, specifically regression modelling, to quantify these factors and their effect on perception. We created a survey to gather the data necessary for this analysis. It consists of three sections: first, background information to help identify the relevant factors for each respondent; next, a "pop" quiz designed to provide insight into the respondent's knowledge of synthetic biology; finally, a section that relates to the respondent's perception of synthetic biology and its uses.
The central question to be answered by this study is "what makes somebody more likely to have a favourable or unfavourable opinion of synthetic biology?" The purpose of this study is to determine the measurable effect of certain factors such as age and field of study or occupation on one's opinion of synthetic biology and its potential applications. This was to be accomplished via a regression model of the form yi = β0 + β1xi1 + β2xi2 +...+ βpxip + εi for i = 1, 2,...n,. Here y represents an individual's perception of synthetic biology, x represents each of the factors considered and β represents the corresponding quantifiable impact, positive or negative, of each factor on perception. Regression analysis was to be conducted using statistical software, most likely Stata or SPSS. The data needed for this analysis was to be collected via an online survey. Respondents will indicate the factors that correlate to them based on their answers to the questions in section #1 of the survey, while section #3 has been designed to reveal the respondent's current perception of synthetic biology. The second section of the survey is a short quiz to illustrate a respondent's level of knowledge and familiarity with synthetic biology. This factor is expected to be the major determinant in perception, along with age range and field of study/occupation. Other factors such as gender and geographic location within Ontario are expected to have no statistically significant impact on perception. The respondents were to come from a wide range of backgrounds in order to increase the robustness of our results.
The rationale behind this study is that by identifying the demographics that are most and least favourable toward synthetic biology and its expanding range of uses, the UW iGEM can more effectively target our efforts for raising awareness on the field. Despite synthetic biology's rapidly growing importance in a wide variety of fields including energy and health, it is still relatively unknown to the population at large. While some may have a vague notion of what synthetic biology is and its potential impact, most do not have anything to associate it with. In fact, some may even find the juxtaposition of artificial (synthetic) and natural (biology) confusing or contradictory. There are even organizations such as the ETC group that have published biased and one-sided reports ("Extreme Genetic Engineering: An Introduction to Synthetic Biology) that are threatening to greatly damage public opinion of synthetic biology. Biotechnology has faced a similar challenge as it has risen to prominence over the past two decades, with misinformation spread and the public lacking the fundamental knowledge necessary to critically interpret this information. In order to combat this reputation, we need to raise awareness of what synthetic biology is, along with an honest and unbiased assessment of its risks and benefits. As this can be a daunting task, the iGEM team decided to conduct this study as a way to help us focus our efforts and gain insight into the composition of perception.
After creating the survey questions, we distributed it online via Kwik Surveys using past co-op employers, campus clubs and other resources. Unfortunately we were not able to accrue enough responses to make any regression analysis statistically significant. Upon meeting with an econometrician in mid-September, we decided to refocus the survey on revealing the mechanism by which some groups end up with specific misconceptions regarding synthetic biology. As an (albeit very exaggerated) example, if the survey were to reveal that respondents with children were much more likely to support the notion that synthetic biology was "playing God by creating life," one might speculate that these respondents feel that as parents only they have exclusive domain of creating life. The value of this past year's project was to gain experience in the areas of survey creation and distribution, as well as to build connections with those who can help take next year's Human Practices to new levels.
TEST TEST page
OUR TEAM!
 Peter Hong
Peter HongDirector

Linda Yang
Assistant Director

Ekta Bibra
Outreach Leader

Anjali Arya
Outreach Leader

Urooj Kishor
Outreach Leader

Angela Biskupovic
Human Practices Leader

Simon Burru
Human Practices Leader

Aaron Bender
Lab Project Leader

Dongbin Zhang
Lab Project Leader

Denise Lieuson
Lab Project Leader

Emily JunWen Li
Lab Project Leader

Rummy Chowdhury
Lab Project Leader
 Kasia Karpinska-Leydier
Kasia Karpinska-LeydierLab Team
 Ashley Ross
Ashley RossLab Team
 Reena Paink
Reena PainkLab Team
 Jordan Lapointe
Jordan LapointeMathematical Modelling Leader

Paul Reginato
Mathematical Modelling Leader

Kathryn Scannell
Mathematical Modelling Team
OUR ADVISORS!
 Dr. Brian Ingalls
Dr. Brian Ingalls Dr. Trevor Charles
Dr. Trevor Charles Dr. Barb Moffatt
Dr. Barb Moffatt Dr. Marc Aucoin
Dr. Marc AucoinUNIVERSITY OF WATERLOO
University of Waterloo was founded in 1957 and has grown to accommodate 30,000 undergraduate and graduate students, and has become Canada's leading university in comprehensive learning. Also, the university has consistently been voted as the most innovative, most likely to produce the leaders of tomorrow, and best overall University in Canada for over 18 years (according to Maclean's Magazine). Waterloo's reputation is however based on its excellent and pioneering co-op program which offers students a balance of work and school on a per term basis, making it a unique learning experience. The city of Waterloo has recognized University of Waterloo and its students, by meeting its demands in terms of funding and involvement. The University has also opened up two new campuses; the pharmacy building, and the joint McMaster medical building in Kitchener, as well as the architecture building in Cambridge, contributing to not only the city of waterloo but the whole Grand River area.
WATERLOO - KITCHENER COMMUNITY
City of Waterloo mainly revolves around the two universities: University of Waterloo and Laurier University. Waterloo is surrounded by Kitchener and thus, the two cities are known as the twin cities, also referred to as Kitchener - Waterloo. The population of the city of Waterloo is always fluctuating due to temporary residents at Waterloo's two universities. Total population in 2009 was recorded to be 121, 700; approximately 20,000 of which were temporary post-secondary students. Due to its small size, people in the past have tried to merge the two cities together but have been unsuccessful. As of today, both cities have their own identity and their own separate city governments.
UW's parts for 2012
BBa_K576003 - RNA - Left part of self-excising ribozyme
BBa_K576004 - RNA - Right part of self-excising ribozyme
BBa_K576005 - Reporter - Left part of GFP (GFP 1) with promoter (J23101) and RBS (B0034)
BBa_K576006 - Reporter - Right part of GFP (GFP 2) with transcription terminator
BBa_K576007 - Intermediate - Left part of GFP with left part of self-excising ribozyme attached using RFC 53 construction.
BBa_K576008 - Intermediate - Right part of the self-excising ribozyme attached to the right part of GFP using RFC 53 construction
BBa_K576009 - Intermediate - Lox attached on to BBa_K576005 on the right of the part. Standard assembly (RFC 10) was used for this construction.
BBa_K576010 - Intermediate - Lox attached on to BBa_K576008 on the left of the part. BBa_K576009 or BBa_K576010 can be used depending on your convenience
BBa_K576011 - Reporter - Final construction of the 2011 project. The self-excising ribozyme should be cut out of from the rest of the sequence and thus expressing the full GFP.
BBa_K576012 - Reporter - Negative control of the experiment. The lox recombination site interrupts the GFP expression
BBa_K576013 - Reporter - Positive control of the experiment. Everything in between has been cut out by the self-excising intron and the GFP is fully expressed.
The UW 2012 lab project is a continuation from the 2011 one, thus no new parts are introduced.

Fig 1. Construction map for experimental main construct
• K576005 contains the first component of GFP (GFPL)
• K576003 contains the first part of the intron sequence (INL)
• J61046 contains the lox site
• K576006 contains the second component of GFP (GFPR)
• K576004 contains the second part of the intron sequence (INR)
• K576007 contains GFPL and INL
• K576009 contains GFPL, INL and lox1
• K576011 contains the promoter (P), ribosomal binding sit (RBS), GFPL, INL, lox site, INR, GFPR and transcriptional terminator (TT). This is the final construct (experimental design)

Fig 2. Construction map for positive and negative controls
• K576005 contains the first component of GFP (GFPL)
• K576006 contains the second component of GFP (GFPR)
• K576013 contains the promoter (P), ribosomal binding site (RBS), GFP and transcriptional terminator (TT). This is the positive control.
• K576005 contains the first component of GFP (GFPL))
• J61046 contains the lox site
• K576006 contains the second component of GFP (GFPR)
• K576012 contains the promoter (P), ribosomal binding site (RBS), GFPL, lox site, GFPR and transcriptional terminator (TT). This is the negative control.
Week of June 17th-24th June 17th: First general meeting with lab volunteers, addressing general lab etiquettes and safety procedures. Explained the general lab working outline and project design. Exchanged contact information and constructed a lab schedule for volunteers and lab leaders.
June 18th: Lab leader (Emily) inoculated parts 118-PSB1C3 backbone(Cmr), 119-InL(Ampr), 120-InR(Ampr), 121-GFPR(Ampr),122-GFPL(Ampr) and 123-Lox (Ampr). All parts except 118 are in puc57, which is ampicillin resistant. Cultures inoculated in LB broth, incubated at 37° C overnight.
June 19th: Volunteers invited into the lab with a brief opening and orientation. Two volunteers trained and conducted miniprep of overnight cultures. Samples are quantified using Nanodrop Spectrophotometer
June 24th: Digested samples (118-122) with Fermentas Fast-digest restriction enzymes EcoR1 and Pst1 to subclone parts 119-122 into PSB1C3 (118). Digestion was incubated at 37° C for an hour (may be too long, but no star activities detected).
A diagnostic gel then follows by loading the cut samples along with the uncut minipreps for comparison.
129bp band observed for InL (119), 172bp observed for InR (120). 545bp and 399bps are observed for GFPL (122) and GFPR (121) respectively. Differences observed between the undigested and digested samples.
June 27th: Run the sample gel as last time but with no control. Gel extracted samples using Biobasic and fermentas binding buffers.
June 28th: Nanodrop results show very low concentration (less than 1uL) for InL and 2. Gel extraction need to be repeated
June 29th: Gel extraction repeated and concentration greatly increased. Enough for ligation
July 4th: Ligation using 1:3 vector to insert ratio in moles. InL, InR, GFPL and GFPR are ligated into PSB1C3 with vector+ ligase control and vector only control. The ligation tubes are incubated overnight at room temperature.
July 5th: Transformation into DH5a cells using heat shock technique. Cells plated on LB Cmr plates and incubated overnight at 37° C. Added transformation control (vector uncut into DH5a) and negative control (DH5a cells only). 1/10 and 9/10 dilutions for each ligation
July 6th: Took out transformation plates. Pink colonies (hundreds) only observed on vector+ligase, vector only controls and transformation controls. Negative control plates are clean. Plates with insert and plasmid have hundreds of white colonies and some pink colonies present. Two morphologies present; large and small
July 9th: Streak purified 24 white colonies selectively (6 of each 3 large, 3 small)
July 10: Inoculate and patched colonies into LB CmR tubes/plates
July 11: Picked 8 final colonies to screen (1 big and 1 small each set) Miniprep these samples
July 12: Digested samples to check for insert (with EcoR1 and Pst1). Loaded a diagnostic gel afterwards
Result: Proper sized inserts were observed for all samples. All parts were subcloned successfully into PSB1C3
July 13: Inoculated for more miniprep
July 14: Miniprep samples again and saved frozen stock of samples
July 18th: Digestion of InL and GFPL with BgIII and EarI (sites overlap, put EarI in first and add BgIII after 30 minutes). Insert (115bp) observed for digestion with InL
July 19th: Gel extracted InL and GFPL
July 21st: Ligated InL with GFPL with 1:3 insert: vector ratio in moles. Incubated at room temperature overnight
July 23rd: Transformed ligation product with transformation and negative control on LB Cm plates
July 24th: All cells had a lawn of cells.
July 26th: Miniprep and digested using the same protocol as above
July 27thh: Miniprep PSB1C3
July 28th: Miniprep lox and digested lox using Xbal and Pst1. Digested lox loaded onto 2% gel.
August 2nd: Gel extracted InL and GFPL again for ligation and conducted ligation again
August 9th: Transformation into DH5a
August 13: Repeat miniprep of InL and GFPL
August 14th: Digested InL and GFPL with EarI and BgIII again
Exam break
August 28th: Final ligation of InL and GFPL
August 29th: Transformation. All colonies are white, but more colonies present for the inesrt than vector + ligase only.
September 17: Picked colonies for inoculation
September 18: Miniprep of samples
September 19: Digestion of samples and loading of gel. No inserts present.
Lab book: Controls
June 21st 2012: Enzyme digestion and gel extraction of pSB1C3, BBa_K576005 in pUC57, BBa_K576006 in pUC57 for subcloning. There were unexpected bands on the gel we will have to redo experiment.
June 29th 2012: Enzyme digestion and gel extraction of pSB1C3, BBa_K576005 and BBa_K576006 for subcloning.
July 7th 2012: Ligation of BBa_K576005 into pSB1C3 and ligation of BBa_K576006 into pSB1C3 for subcloning.
July 10th 2012: Transformation of BBa_K576005 in pSB1C3 and BBa_K576006 in pSB1C3 into DH5-alpha for subcloning.
July 11th 2012: Checked the plates and they were all lawns. We will use Emilys subcloned parts for now.
July 17th 2012: Inoculation of pSB1C3 containing strain and BBa_J61046 in pUC57 containing strain.
July 18th 2012: Our frozen stock may be contaminated so we discarded the inoculation tubes from July 17th and we will use Emilys miniprepped samples for now. Restriction digest and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006.
July 21st 2012: Ligation to make BBa_K576013 construct.
July 22nd 2012: Transformation of BBa_K576013 construct into DH5-alpha.
July 23rd 2012: All the plates were lawns. Shaking the broth we used showed it was contaminated. Discard the plates and repeat the experiment.
July 28th 2012: Restriction digestion and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006. The bands on the gel looked as expected but the nanodrop values after the gel extraction were indicated we did not have DNA. We will repeat the digest and gel extraction using isopropanol to increase yield.
August 18th 2012: Restriction digestion and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006. Ligation to make BBa_K576013 construct.
August 19th 2012: Transformation of BBa_K576013 construct into DH5-alpha. Transformation of left construct intermediate for the main construct.
August 20th 2012: All the plates were lawns. This time we are confident the broth was not contaminated. Our problem could be with the antibiotic. Next time we will plate a 1/10 dilution of the cells.
August 21st 2012: Restriction digestion and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006. The bands on the gel looked as expected but the nanodrop values after the gel extraction were not good enough to move onto ligation.
August 26th 2012: Restriction digestion and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006. Restriction digestion of pSB1C5. pSB1C5 showed an unexpected band in the gel so we did not proceed to gel extraction.
August 28th 2012: Ligation to make BBa_K576013 construct.
August 29th 2012: Transformation of BBa_K576013 construct into DH5-alpha. Transformation of left construct intermediate for main construct into DH5-alpha.
August 30th 2012: All the plates were clean except the transformation of the miniprepped sample for a positive control had colonies. Our problem is therefore not with the transformation. Our problem may be with our ligation. We will repeat the experiment with an additional control for the ligation.
September 6th 2012: Restriction digestion and gel extraction of BBa_K576005 in pSB1C3 and BBa_K576006. Linearize and gel extract pSB1C3 for additional ligation control. Ligation to make BBa_K576013 construct and ligation of linearized pSB1C3.
September 7th 2012: Transformation of BBa_K576013 construct into DH5-alpha.
September 8th 2012: The transformed miniprep and transformed ligated pSB1C3 had colonies. The rest of the plates were clean. Therefore the transformation and ligation was successful however the construct still didn't work out. We will repeat the experiment after miniprepping more BBa_K576006 in pSB1C3. Inoculate BBa_K576005 in pSB1C3.
September 9th 2012: Miniprep BBa_K576006 in pSB1C3.
September 11th 2012: Streak purify DH5-alpha carrying BBa_K576005 in pSB1C3, DH5-alpha carrying BBa_K576005 in pSB1C3 and DH5-alpha carrying BBa_J61046 in pUC57 from frozen stock.
September 12th 2012: Inoculate DH5-alpha carrying BBa_K576005 in pSB1C3, DH5-alpha carrying BBa_K576005 in pSB1C3 and DH5-alpha carrying BBa_J61046 in pUC57 in Terrific broth.
September 13th 2012: Miniprep BBa_K576005 in pSB1C3, BBa_K576006 in pSB1C3 and BBa_J61046 in pUC57.
September 15th 2012: Restriction digestion of parts BBa_K576005, BBa_K576006 and BBa_J61046 to get gel extracted fragments for both the positive and negative control constructs. Diagnostic gel showed questionable bands so we need to repeat the digestion.
September 16th 2012: Repeat the digestion from September 15th. This time the bands are as expected so the fragments were gel extracted. The nanodrop values of gel extracted BBa_K576005 in pSB1C3 for the negative control construct indicated we did not have DNA so need to redo that sample. Other samples were stored.
September 18th 2012: Restriction digestion of BBa_K576005 in pSB1C3 for negative control construct but the gel showed an unexpected band so did not gel extract. Ligation to make BBa_K576013 construct. Inoculation of DH5-alpha carrying BBa_K576005 in pSB1C3, DH5-alpha carrying BBa_K576006 in pSB1C3, DH5-alpha carrying BBa_K576003 in pSB1C3, DH5-alpha carrying BBa_K576004 in pSB1C3 and DH5-alpha carrying BBa_J61046 in pUC57 in Terrific broth.
September 19th 2012: Miniprep of BBa_K576005 in pSB1C3, BBa_K576006 in pSB1C3, BBa_K576003 in pSB1C3, BBa_K576004 in pSB1C3 and BBa_J61046 in pUC57. Transformation of BBa_K576013 construct into DH5-alpha.
September 20th 2012: All plates were clean except the transformed miniprep. Stop lab work on BBa_K576013 construct due to time constraints.
September 23rd 2012: Restriction digestion of minipreps from 1-12 colonies to screen left construct transformation plate. Load diagnostic amounts of digested samples to image BBa_K576005, BBa_K576006, BBa_K576003, BBa_K576004 and BBa_J61046 bands in a gel to demonstrate successful subcloning. The BBa_K576003 and BBa_K576004 bands were not visible in the gel.
September 24th 2012: Load diagnostic amounts of digested miniprepped 1-10 colonies to screen left construct plate.
SAFETY
Laboratory Safety
The Ribozyme Project is not expected to raise any research, public or environmental safety concerns other than those normally associated with Biosafety Level 2 organisms, such as Escherichia coli (DH5-alpha), which is classified as very low to moderate. The use of this project is primarily reserved for research and laboratory use, therefore, should not purposefully be exposed to the public or environment except after further testing in its specific applications (such as with particular fusion proteins). Furthermore, the basis of our project is to establish a self-excising sequence (ribozymes), which should limit the expression of any intervening sequences to the RNA level. If the intervening sequence were something of environmental or public relevance (such as antibiotic resistance), the experimental design indicates that the sequence will be removed and, thus, not expressed. This is a relevant contribution of the design in limiting expression to the RNA level, which eases environmental hazard concern upon the accidental release of a GMO containing this biobrick. Therefore, the new biobrick parts submitted should not raise any safety issues.The necessary facility, equipment and handling procedures associated with Level 2 Biosafety concerns were met:
1.Pipetting aids
2.Biosafety cabinets where applicable
3.Laboratory separated from other activities
4.Biohazard sign
5.Proper safety and disposal equipment, including autoclave
6.Personal protective equipment, worn only in the laboratory
7.Screw-capped tubes and bottles
8.Plastic disposable pasteur pipettes, when necessary
All precautions with respect to recombinant DNA were observed:
1.All waste was autoclaved before being thrown away.
2.Researchers practiced aseptic technique and personal hygiene and safety precautions
3.Procedures likely to generate aerosols are performed in a biosafety cabinet
4.Bench surfaces were disinfected with ethanol.
4.Potentially contaminated waste is separated from general waste
Safety Questions
1. Would the materials used in your project and/or your final product pose: The materials used in the lab are non toxic to health of individuals as well as to the environment. One of the major reagents that is used is GelRed which is used as a substitute for Ethidium Bromide. Gel Red is unable to penetrate into cells and so is a non-mutagenic agent. As well it has the same spectral characteristics as Ethidium Bromide and so has the same effectiveness of use. The project itself is safe even if released into the environment by design or accident since the part being expressed is the Green Fluorescent Protein (GFP). Unless the sequences are mutated, the project poses no risk.
Please explain your responses (whether yes or no) to these questions.
Specifically, are any parts or devices in your project associated with (or known to cause):
- pathogenicity, infectivity, or toxicity? No
- threats to environmental quality? No
- security concerns? No
The parts that are associated with the project this year are at the same level of risk as the any of the regular parts that already exist. All parts are constructed in an antibiotic containing backbone so that accidental release of will pose minimal risk to contaminating other bacterial populations.
2.Under what biosafety provisions will / do you operate?
a.Does your institution have its own biosafety rules and if so what are they? The University of Waterloo had a Bio-Safety plan in place to ensure the proper use to bio-hazardous materials in teaching and research at the university. A more detailed overview of their plans is outlined at the Bio-Safety Website
b. Does your institution have an Institutional Biosafety Committee or equivalent group? If yes, have you discussed your project with them? The laboratories operating at the University of Waterloo have obtained permits from the Bio-Safety Committee in order to perform intended research. Since the Waterloo iGEM team performs all laboratory work in a parent lab under the guidance of the Masters and PhD students of that lab, the projects carried out in the lab are covered by the permits obtained by the parent lab.
c. Will / did you receive any biosafety and/or lab training before beginning your project? If so, describe this training. All lab volunteers are required to take an online training to familiarize themselves with the Biosafety practices of the University of Waterloo. The training is followed up by a quiz ensuring proper understanding of the material. Upon completion of the training and quiz a hands- on lab training is provided under supervision of the parent lab's PhD student. The hands-on training involves instruction of use of the appropriate equipment that is used in the lab, as well as how to maintain and discard materials in a safe manner.
d. Does your country have national biosafety regulations or guidelines? If so, provide a link to them online if possible. Canada operates under the guidelines set up by the Public Health Agency of Canada. The Agency is the national authority on matters concerning biosafety and biosecurity. Risks to the public are reduced by standardizing controls over activities that involve human pathogenic agents, domestic or imported. While these guidelines are in place the current iGEM project does not involve work with any agents or materials that may pose a risk to humans. The link to the Public Health Agency of Canada is provided below: Public Health Agency of Canada
