 "
"
Team:MIT/Results
From 2012.igem.org


RNA Strand Displacement In Vitro
Previously:
In 2011, Lulu Qian and Erik Winfree, researchers at Caltech, published a paper entitled "Scaling Up Digital Circuit Computation with DNA Strand Displacement Cascades." This paper demonstrated how scalable logic circuits based on DNA strand displacement are capable of processes as complicated as the square root function. See our motivation page for more details.
MIT iGEM 2012:
Before our team attempted to bring the mechanism of strand displacement into an in vivo context, we first decided to assay strand displacement in vitro using RNA. We used 2'-O-methylated RNA strands, which had not been shown to undergo strand displacement in vitro. Before creating our own constructs, we adapted sequences from the Qian/Winfree paper to RNA.
MIT iGEM Foundational Experiment:
Figure A shows a foundational in vitro RNA strand displacement experiment that was performed on a plate reader. The negative control, in black, is a well that received only an annealed reporter complex. The bottom strand of this complex is the gate strand, T*-S6*, with the 3' end tagged with the ROX fluorophore. The top strand of the complex is the output strand, S6. This is complementary to the S6* domain of the gate strand. The 5' end is tagged with the Iowa Black RQ quencher, which absorbs the ROX fluorescence; thus, when the two strands of the reporter are annealed, no fluorescence should be observed. The positive control, in red, is the input strand, T-S6, annealed to the gate strand, T*-S6* tagged with ROX. This is what we would expect the product of a strand displacement reaction to look like. We can see that in the experimental well, when the input is present, it can bind to the exposed T* domain of the reporter and displace the output strand, yielding a fluorescent complex and a waste strand.
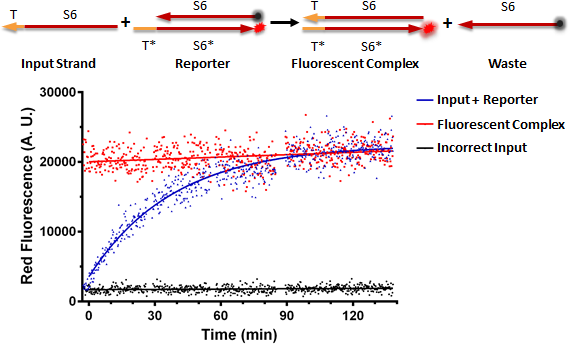
Figure A
Nucleic Acid Delivery
In order to implement RNA strand displacement cascades in vivo, we first demonstrated our ability to deliver nucleic acids to mammalian cells. We have achieved the delivery of plasmid DNA, single-stranded modified RNA and double-stranded modified RNA to mammalian cells through both lipofection and nucleofection.
(1) Delivery of Plasmid DNA to Mammalian Cells
Through the Gateway method, we have assembled many promoter-gene constructs as detailed on our Parts Page. After construction, we deliver the plasmid DNA to Mammalian Cells through the use of transient transfection, lipofection with Lipofectamine 2000 reagent. Figure A shows:
Figure A will go here.(2) Delivery of 2'-O-Me RNA to Mammalian Cells
Since our modified reporter constructs use 2'-O-Methyl RNA, we must be able to deliver 2'-O-Me RNA into mammalian cells. The movie below shows HEK293 cells expressing constitutive eYFP with a 2'-O-Methyl RNA strand labeled with ROX (5-carboxy-x-rhodamine) on the 3' end. As time passes, the complex/vesicles are uptaken by the cell, releasing their payload resulting in whole cell fluorescence. Each frame is 5 minutes, movie encompasses 200 minutes in 9 seconds.
Delivery of ROX-labeled 2'-O-Methyl RNA into HEK cells.
![]() Time point images taken at t = 0, 2, 3, and 4 hours post-transfection. Images taken at 10X on Zeiss microscope.
Time point images taken at t = 0, 2, 3, and 4 hours post-transfection. Images taken at 10X on Zeiss microscope.
Once we demonstrated ability to deliver 2'-O-Me RNA to mammalian cells, we ran optimization experiments to optimize the ratio of RNA delivered to transfection reagent used. 2'-O-Me RNA to transfection reagent.
Figure to be added - 15,20,25,30 pmol ratio DATA from FACS.
(3) Inducible Control of Protein Expression

Caption needed.

Caption needed.
Delivery of Plasmid DNA which transcribes short RNA Inputs
FF1 Knockdown Data with triplicate data from nathan
In Vivo RNA Strand Displacement
Strategy 1: Lipofectamine 2000 Transfection of RNA version of Reporter from Winfree/QIan 2011 Paper

Caption needed.
Images will go here from April 24th experiment - display red fluorescence in all wells, including those that only got reporter or the wrong input - also see red vesicles indicating reporter comes apart inside the vesicles
Strategy 2: Switch Transfection reagent to RNAiMAX
RNAiMAX is supposed to be a better transfection reagent for double stranded RNA
Images will go here from experiment from June 13th onward where we do not see red vesicles, however we still see whole cell red fluorescence
Strategy 3: Tag RNA strand with an Alexa Fluorophore to act as a transfection marker
Strategy 4: Create DNA plasmids driving transcription of RNA inputs, while transfecting RNA Reporter
Strategy 4: Nucleofect RNA reporter, RNA inputs
[Strategy 5]: Redesign RNA Reporter
Sensing Overview
We designed, modeled and tested an mRNA sensor that interfaces with RNA-based strand displacement circuitry.
Sensing Design
We imposed the following criteria on our mRNA sensor design:
- orthogonality
- easy integration with strand displacement circuits
- ability to amplify a signal
- ease of sensor generation
The way we implemented this sensor is by considering the mRNA sequence and choosing within it two consecutive abstract domains: the toehold and the sensing domains. These domains mirror the toehold and recognition domains in strand displacement, so provide an interface to any strand displacement circuit. Specifically, the chosen toehold and sensing domain will then directly interface with an intermediate gate:output complex using toehold-mediated strand displacement, with signal amplification achieved using a fuel strand.

Illustration of an mRNA sensor. A toehold region in the mRNA can bind to the toehold on a gate:output complex. Branch migration can then occur, resulting in a free output signal. Using fuel, the mRNA can be released from the gate, enabling it to react with another gate:output complex.
However, this abstraction needs to take into account the secondary structure of mRNA: some regions are more accessible (less basepairing), while others are strongly basepaired and thus unavailable to be directly sensed by mechanisms that involve basepairing to the mRNA (Kertesz et al. 2007).

Software rendered secondary structure of eBFP2 (BBa_K779300) showing various secondary structure elements (e.g. stems and loops)
Much work has been done to generate these secondary structures computationally (see nupack.org), and to find accessible regions predicted to be miRNA binding sites within mRNAs (e.g. PITA). We leveraged these algorithms to identify potential toehold and sensing domains within mRNAs by looking for regions with different levels of accessibility.
Once suitable domains have been chosen using modeling of the secondary structure, we can rank them by their orthogonality to other transcribed RNAs. These sequences are available in online databases (e.g. mRNA data for HEK 293 cells). For strand displacement, the rate limiting step is the binding of a strand to a gate:output complex using the toehold. After this step, the process of strand displacement is sensitive to nucleotide mismatches, with early mismatches being more disruptive than later mismatches (Qian et al. 2011). Thus, we can use a weighted Hamming distance between a candidate domain and every subsequence in the transcriptome. This method is similar to identifying orthogonal sequences for protein-DNA interactions as previously published (Silver et al. 2012).
Care has to be taken to make sure that sensors in a circuit are orthogonal as well, but the same method can be applied to any sequences that are introduced into a cell.
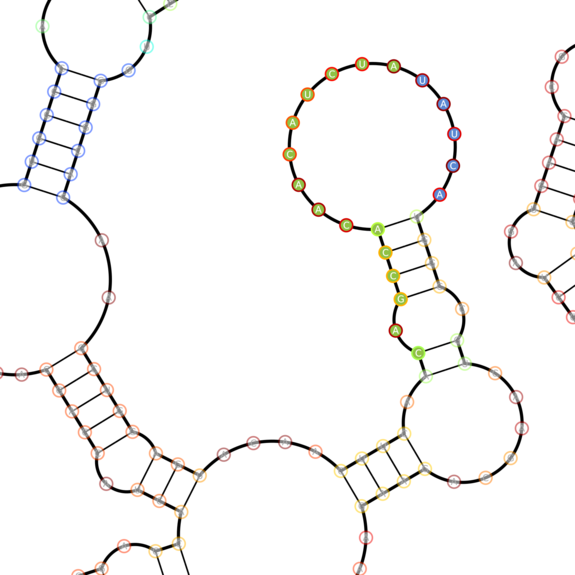



Four highlighted domains of eBFP2 with various secondary structures we chose to sense. Blue-filled circles represent toehold nucleotides, green-filled nucleotides represent sensing domain nucleootides. The border of a nucleotide's circle represents the probability of the nucleotide being in the given state (red - very likely, blue - very unlikely). These are closeups of the mRNA structure above.
Sensing Modeling
Modeling of mRNA secondary structure was done using NUPACK. Strand displacement reaction kinetics were simulated using Visual GEC.
Sensing In vitro studies
We chose 4 domains in eBFP2 with various predicted secondary structure properties to test out in vitro. eBFP2 (BBa_K779300) mRNA was produced by PCRing on a T7 promoter and terminator (TODO: link to primers?). The resulting template was then used for in vitro transcription (see Protocols TODO: link). After purification and quantification on a NanoDrop 1000, the transcribed mRNA, corresponding gate:outputs and fuel strands along with a fluorescent reporter were added to wells in a 96-well plate and the fluorescence was measured on a plate reader (See TODO: link to protocol).
For proof-of-concept studies we chose DNA as nucleic acid for the gate:output, fuel, and reporter complexes, as these results will mirror results with RNA-based strands, as shown by foundational experiments. However, the thermodynamics, kinetics and steady states will be different between DNA and RNA strands. We expect mRNA to produce less output than a corresponding DNA input mimic (comprising of just the toehold and sensing domains) in the same amount.
Results indicate that there is a difference between the 4 domains we chose, and that the output signal from mRNA is less than the signal from DNA mimics.

In vitro results from a DNA-based mRNA sensor. Graphed here is the fold increase in RFU - comparing the pre-input fluorescence to the fluorescence after (TODO) hours. Baseline (light gray) reflects no input. DNA input mimic (dark gray) is a DNA oligonucleotide with the same sequence as the toehold and sensing domains in mRNA (black).

Comparing mRNA as an input to DNA as an input. Graphed here is the ratio of fold increase fluorescence comparing mRNA inputs to DNA inputs. This indicates the possible completion level due to mRNA inputs. As described above, we expect this to be <1.
Not Gate in vitro

Figure 1 - caption needed
One of the possible application of the in vivo RNA strand displacement is to sense high and low concentration of specific biomarkers to distinguish, for instance, healthy cells from cancerous cells. The sensing part of our circuit will translate biomarkers in the form of mRNA in short strand, non coding, RNA. These short strands will be the input of the processing part of our circuit. To perform correctly the needed information processing, we need to 'transform' specific low signals (that is, low concentration of short strands non coding RNA) in high signals that then can be processed by a downstream AND gate. This transformation can be obtained by a NOT gate, where the input and output are the above-mentioned short strands RNA.
We first implemented the NOT GATE in vitro using DNA instead of RNA strands. The design of this gate is in figure 1, where a letter with a '*' depicts a complementary domain to the one denoted by the letter alone. We arrived to this design after having conceived other 5, trying each time to reduce the number of molecules involved or their complexity.
To understand the behavior of this NOT GATE it can be useful to consider two extreme cases, that is, no input present and input at high concentration.
When the input is not present the B molecules can bind reversibly with A and reversibly with C. When B start to displace c2 from C, the D molecules will free B that consequently will be able to displace c2 from other C molecules. Finally c2 will irreversibly displace e2 from the readout. Therefore we will see high fluorescence (that is, high level of output with no input)
When the input is present in high concentration, B and the input bind irreversibly with A due to the mechanism of the cooperative hybridization(Cooperative Hybridization of Oligonucleotides,David Yu Zhang,JACS 2011) , therefore B cannot displace anymore c2 from C. Consequently e2 cannot be displaced from the readout. Therefore we will see no fluorescence (that is, no output with high level of input).

Experimental result for the in vitro NOT GATE where the output fluorescence is normalized to the highest value of the NOT GATE transfer function and the total volume for each level of input was 100ul.
The relative concentration of A with respect of input and B is extremely important. Indeed if the concentration of A is too low the cooperative hybridization between A , B and a high concentration of input can be slow, consequently B can displace c2 from C, that is, we would have a high level of output although the input level is high. On the other hand if the concentration of A is too high, even without the presence of input, B will continuously reversibly bind with A. Consequently B will not displace c2 from C and therefore we would not see a high level of output when the level of input is low.
In addition to the relative concentration of the different components another important point is the absolute concentration of them. This is mainly due to how the cooperative hybridization works. Indeed the reactants are three and the products two, consequently at low concentration the reactants are more favorable in the reaction whereas at high concentration the products will be more favorable.
Our strategy consisted first in finding the right concentration to let the cooperative hybridization works and then we tuned the concentration of A to find the right trade of between the interaction of A, input and B when the input is high and the interaction of A and B when the input is low.
Decoys and Tough Decoys (TuDs)
We wanted something that would provide a tight double repression system with a very distinct change between on and off. The TuDs and Decoys design were originally inspired by the “Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells,” paper. We copied their designs and wanted to reproduce the results in our lab. To do so, we ordered TuDs and decoys both with and without bulges. The bulges are designed to disrupt RISC complex activity; something which degrades short RNA like our decoys in the cell.
Sources: http://nar.oxfordjournals.org/content/early/2009/02/17/nar.gkp040.abstract
http://www.ncbi.nlm.nih.gov/pubmed/9695408