 "
"
Team:Virginia/Project
From 2012.igem.org

Contents |
Project Overview
Whooping cough, the infectious respiratory disease caused by Bordetella pertussis, is diagnosed in tens of millions of people and results in almost 300,000 deaths globally each year. Low-income and unvaccinated individuals as well as infants are especially susceptible. Current diagnostic procedures are complicated, costly, and can take up to a week, by which time the disease may have progressed or spread. The enormous impact of this disease urgently motivates the development of a faster, cheaper, and more reliable diagnostic. Our epidemiology models suggest that earlier diagnosis could drastically reduce the incidence and impact of the disease. We propose an engineered bacteriophage diagnostic system for rapid clinical detection of pertussis.
As a proof of principle, we worked on engineering the T7 bacteriophage to express a version of the beta subunit of human chorionic gonadotropin (hCG) which we codon-optimized for expression in E. coli. Due to host-pathogen specificity, bacteriophages will only replicate and produce intracellular hCG if the target bacteria is present in the sample. The bacteriophage lyses the cell, releasing the hCG, which can then be detected using a pregnancy test. Pregnancy tests are commonly available in all clinics and can detect very low concentrations of hCG, which is an advantage over currently-available methods for detecting phage amplification. The bacteriophage system also requires minimal training to be used in lab and does not require any high-tech machinery. This will make the diagnostic readily usable in developing countries, where whooping cough is a particular concern. Besides pertussis, this method can be used to detect the presence of any bacteria for which there exists a bacteriophage using a common pregnancy test.
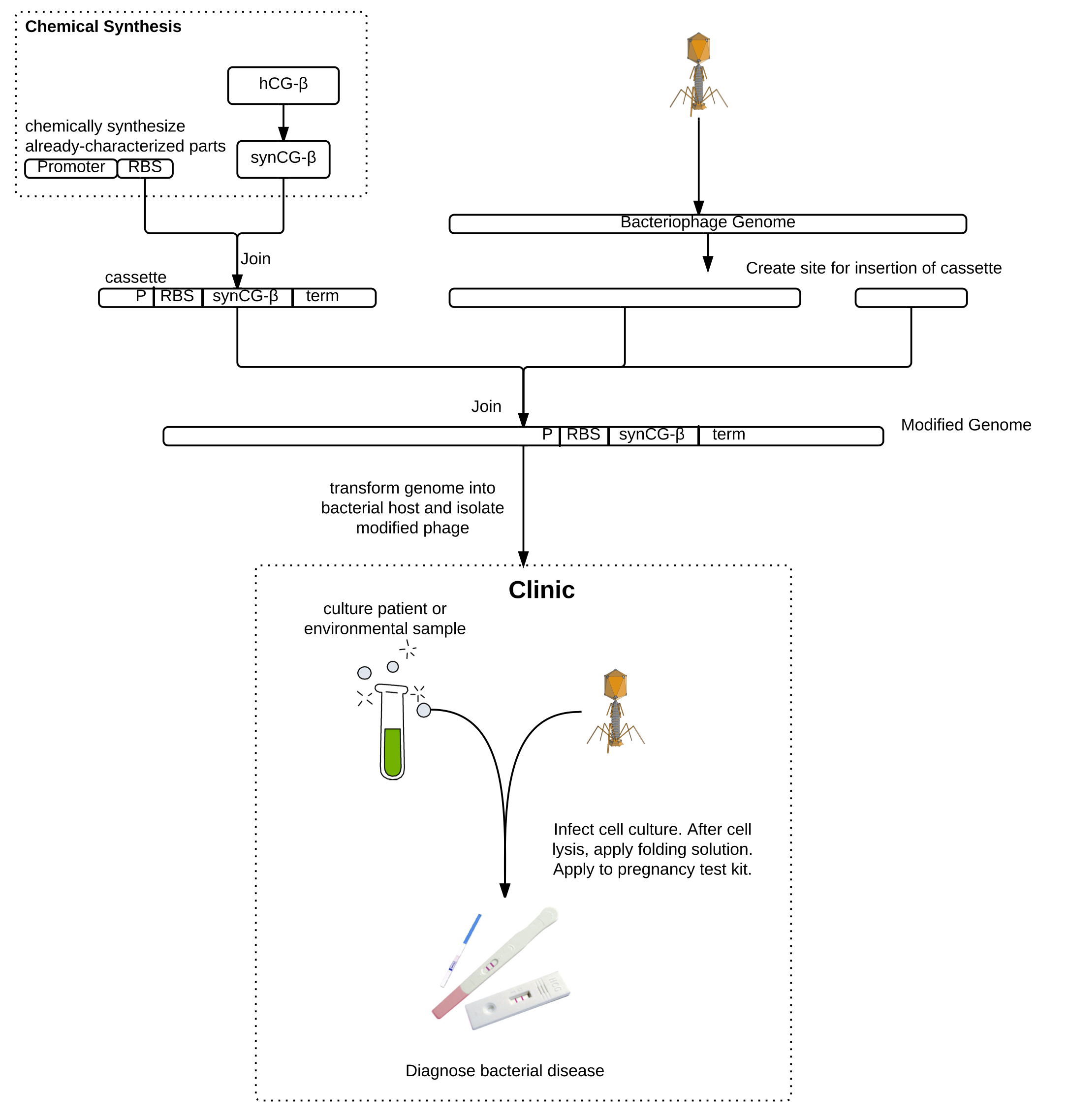
Background
Limitations of Current Detection Methods
To diagnose whooping cough, doctors are currently limited to observing symptoms and detecting the causative bacteria via problematic laboratory tests: PCR, culturing, or serology. Because early symptoms are non-specific, differential diagnosis difficult. PCR is rapid and sensitive, but is highly prone to false positives, can be triggered by vaccines, and requires expensive lab equipment and expertise. Culturing from nasopharyngeal samples can achieve up to 100% specificity, but performing a culture analysis can take several days to a week, and sensitivity decreases with symptom duration. Additionally, culturing sensitivity decreases rapidly after the first two weeks of the cough. Finally, serology can be used to confirm diagnosis between 2 and 8 weeks after cough onset during suspected outbreaks, but is difficult to use for diagnosis in vaccinated and adult individuals.
Bacteriophage Basics
Bacteriophage are viruses which specifically infect bacteria, hijacking the bacteria's own replication machinery to replicate themselves. Due to host-pathogen co-evolution, bacteriophage are often extremely specific to a particular genus, species, or even strain of bacteria. They may undergo the lytic cycle, in which the genome of the bacteriophage is used to produce new bacteriophage which are released from the bacterial cell following cell death and lysis, or the lysogenic cycle, in which the bacteriophage genome replicates along with the host genome but does not produce new viruses.
Bordetella Phage
Bacteriophage BPP-1 infects the genus Bordetella, which includes the human pathogen B. pertussis and its cousin B. bronchiseptica. It normally binds to the cell-surface receptor pertactin, which is only expressed during the Bvg+ phase of the Bordetella life cycle (during which the bacteria expresses virulence and colonization factors at high levels). However, BPP-1 also possesses a diversity-generating retroelement system which allows it to switch its tropism (Liu et al., 2002) (Liu et al., 2004).
T7 Phage
T7 is a lytic phage that infects E. coli by binding to lipopolysaccharides on the host cell membrane and injecting its DNA. The T7 genome rapidly makes accommodations to replicate itself, turning off host genes for restriction enzymes, inhibiting nucleases and native polymerases, and redirecting metabolic resources toward its own use. The genome then undergoes replication and concatemerization, and produces about hundred progeny before the host cell is lysed in under half an hour (Qimron, 2010).
Human Chorionic Gonadotropin
Human chorionic gonadotropin is a naturally occurring glycoprotein in the human body, produced in the placenta during pregnancy. It has an alpha and a beta subunit, which are 92 and 145 amino acids long, respectively. In vivo, the large amount of oligosaccharide chains are highly glycosylated, and this highly glycosylated version of the protein is in a great proportion to unmodified hCG in early pregnancy because it is produced by cytotrophoblast cells at implantation of the egg (Cole, 2003)(Cole, 2006). The primary physiological function of the unmodified protein is known to stimulate progesterone manufacture by the corpus luteal cells (Cole, 2003), whereas data on the glycosylated version suggests that it functions in signaling the growth of cytotrophoblast cells and placental implantation (Cole, 2010).
Pregnancy Tests
The pregnancy test is an immunoassay, first developed in 1989 and has since expanded to production by 40 different producers, including several large companies such as INSIGHT pharmaceuticals and Procter & Gamble’s ClearBlue (Cole, 2003)(Canfield, 1989). Like in the first immunoassay developed, most pregnancy tests consists of an antibody against the beta-subunit of the dimer hCG that is immobilized on the test by a secondary antibody. When solution containing the beta-subunit of hCG is dropped onto the test, it is wicked through the immunoassay and causes a chemical change to indicate the presence or absence of the protein.
Methods
Sequence Design
Primers were designed with the aid of PRIMER3. The sequence of synCGβ was codon-optimized in Gene Designer for expression in bacteria based on the the nucleotide sequence of hCG-β from NCBI.
Isolation of Phage and Phage Genome
Phage genomic DNA was isolated from purified phage using the QIAamp Viral RNA kit, which is suitable for isolation of viral RNA and DNA. Phage was amplified, titered, and isolated using protocols which are available on our OpenWetWare page.
Cloning
Plasmid from the BioBrick distribution kit containing the pSB1C3 backbone was transformed into chemically competent DH5α E. coli using a 20 second heat shock at 42°C. Transformants were grown on LB/chloramphenicol (20μg/μl) plates overnight, and plasmid was isolated from inoculations of colonies. Restriction digests were carried out using NEB enzymes and protocols, and the digested product was isolated by either QIAquick PCR cleanup kit or ethanol precipitation. In order to reduce the chance of self-ligation, 5’ ends of the backbone were dephosphorylated by calf intestinal alkaline phosphatase (CIAP) treatment (Promega protocol) and cleaned up.
BioBrick inserts were prepared similarly, with restriction digests and clean up; however, some inserts underwent initial PCR amplification to add BioBrick prefixes and suffixes, and no inserts were CIAP-treated. Ligations were carried out (3 hours at room temperature with T4 DNA ligase from NEB) using various ratios of vector:insert, and immediately used to transform chemically competent DH5α. Transformants were plated, colonies were grown up, and plasmid was isolated, PCR-amplified, and run on agarose gel to confirm presence of insert.
An additional cloning step was carried out to insert wild type hCG-β into the pET-30a vector under the control of the T7 promoter and lac operator. The resulting recombinant gene included a C-terminal His-tag. This plasmid was used for initial characterization of hCG-β.
Phage Modification
Our approach to modification of Bordetella phage used Gibson assembly, a one-step reaction in which 5’ ends are chewed back by 5’ exonuclease, complementary overhangs base-pair and are extended by DNA polymerase, and then gaps are sealed by DNA ligase (Gibson, 2009). Gibson assembly master mix was purchased from NEB, and the reaction was run at 50°C for 1 hour. Subsequent PCR and gel confirmations revealed that the reaction products were a heterogeneous mixture of original and modified genome.
Chemically competent BL21pLys E. coli were transformed with wild type T7 phage genome, incubated at 37°C, monitored spectrophotometrically at O.D.600 for several hours. Chemically competent Bordetella bronchiseptica were transformed with BPP-locked genome and monitored for O.D. as well.
Folding Solution
Before attempting to detect hCG-β produced from modified bacteriophage, an experiment was conducted using recombinant His-tagged hCG-β in pET-30a vector. Cells were induced with IPTG (1mM) for several hours, lysed by various methods (either B-PER, T7 phage, or sonication), and exposed to denaturing and refolding conditions. These conditions were adapted from a protocol used by Huth et al.
Lysis by B-PER (bacterial protein extraction reagent from Thermo Scientific): cells were washed with BPER and incubated for 10 min. Lysis by T7 bacteriophage: bacterial culture in log phase (45 mL, OD~0.650) was infected with purified phage (50μl) and incubated (37°C, 2 hours shaking), after which OD dropped to 0.1-0.2. Lysis by sonication: bacterial culture was sonicated for six 1-minute intervals interspersed by 1-minute resting periods.
Following lysis, three denaturing conditions varying in the reducing agent DTT (0, 0.5, and 5mM DTT) and a fourth condition without denaturing solution were applied. Cell lysates were spun down (12,000g, 10 min), and pellets were collected and resuspended in Tris buffer (1mL 50mM). Resuspended cells (200μL) were aliquoted to either denaturing or non-denaturing (control) solution (800μl). The denaturing solution contained 6M, 50mM Tris (pH 8.9), and variable DTT. The control solution did not increase urea concentration. Lysate/denaturing mixtures were incubated (1 hour, 37°C). As a negative control, some mixture was applied dropwise to a pregnancy test kit, as per instructions on the kit.
After incubation, each mixture was titrated with folding solution in order to render hCG-β detectable by a pregnancy test kit. Folding solution was prepared so that upon its addition, final concentrations of reagents were 4.8mM Cysteamine, 4.4mM Cystamine, 1M Urea, and 1mM EDTA. Titration with folding solution (0.5ml/minute for 14 minutes) brought the final volume to 8mL.The final mixture was incubated at room temperature for 16 minutes and applied dropwise to pregnancy tests. Test kits remained at room temperature overnight and were read the following day.
Results
Phage Production
Following transformation, and after four hours of incubation during which cell density increased, the O.D. of the E. coli transformants steeply declined. Clearing was also observed upon addition of the transformants to a fresh culture of E. coli. However, no clearing was observed when T7 genomic DNA was mixed with non-competent cells, demonstrating that the previous results were indeed due to transformation, and the genome was not contaminated with intact phage particles. These results support that T7 phage genome was taken up by host cells, transcribed and translated, and packaged into intact, infective phage. Production of BPP phage was not observed in transformations with bronchiseptica, so the Gibson assembly product was not used to produce modified BPP phage.
BioBrick Confirmation

Gel electrophoresis of PCR-amplified regions of potential BioBricks. The optimum lanes are WT hCG lane 2 and synCG lane 1; the samples that were used to generate these products were submitted to the Parts Registry.
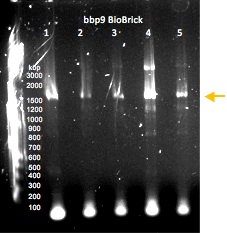
Gel electrophoresis of PCR-amplified regions of potential BioBricks containing the lysin gene bbp9. The sample that was used to generate the product in lane 4 was submitted to the Parts Registry.
Pregnancy Tests + Folding Solution

Lysis of cells expressing wild-type hCG in the pET-30a vector with B-PER reagent initially resulted in no positive pregnancy tests. When the pregnancy tests were checked the next morning (>12 hours after the initiation of the experiment), there was one positive band on the pregnancy test treated with 17-hour induced whole cell extract with no denaturant. We suspected that the B-PER was interfering with the ELISA and switched to lysis with T7 and sonication.

We then attempted T7 lysis and sonication using a culture expressing the synCG device. Applying a denaturing solution without DTT to a combination of four synCG cultures yielded a dark stain on the entire left side with a darker band in the test region, for both T7 lysis and sonication.
T7 modification
T7 phage genome has been isolated, restricted at a unique BclI site, cleaned-up, CIAP-treated, and cleaned up again. The genome is in two 5’-dephosphorylated pieces and ready for ligation with an insert. This unique restriction site was used by Lu and Collins (2007) to insert a gene encoding a biofilm-degrading enzyme into T7. We are in the process of inserting synCG under the control of a constitutive T7 promoter and strong RBS and ending in a hairpin terminator. Once ligated, BL21 chemically competent cells will be transformed with the modified genome. After a drop in OD, signifying the production of phage, the modified phage can be amplified, isolated, and used to detect E. coli.
Conclusions
We propose a novel diagnostic for bacterial disease using engineered bacteriophage and pregnancy tests. We demonstrated that the synthetic version of hCG we designed, in the composite device we constructed to express it, successfully produces hCG detectable by $1 pregnancy tests in bacteria. Next steps include completing the insertion of our construct into phages, optimizing the hCG-folding solution to be used clinically, and expanding our approach to the detection of pertussis and other organisms.
References
Canfield, R. E., Armstrong, E. G., Ehrlich, P. H., & Birken, S. (1989). U.S. Patent No. 4,851,356. Washington, D.C.: U.S. Patent and Trademark Office.Chan, L. Y., Kosuri, S., & Endy, D. (2005). Refactoring bacteriophage T7. Molecular Systems Biology. 1(18). Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/16729053.
Cole, L. A. (2012). The hCG assay or pregnancy test. Clinical Chemistry and Laboratory Medicine. 50(4), 617-30. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/22149742.
Cole, L. A., Daia, D., Butler, S. A., Leslie, K. K., & Kohorn, E. I. (2006). Gestational trophoblastic diseases: 1. Pathophysiology of hyperglycosylated hCG. Gynecologic Oncology. 102(2), 145-150. Retrieved from http://www.sciencedirect.com/science/article/pii/S0090825806002253.
Cole, L. A., Khanlian, S. A., Sutton, J. M., Davies, S., & Stephens, N. D. (2003). Hyperglycosylated hCG (invasive trophoblast antigen, ITA) a key antigen for early pregnancy detection. Clinical Biochemistry. 36(8), 647–655. Retrieved from http://www.sciencedirect.com/science/article/pii/S0009912003001085.
Cornelis, P. (2000). Expressing genes in different Escherichia coli compartments. Current Opinion in Biotechnology. Volume 11(5), 450-454. Retrieved from http://www.sciencedirect.com/science/article/pii/S0958166900001312.
Dublanchet, A. (2007). The epic of phage therapy. Canadian Journal of Infectious Diseases & Medical Microbiology. Bourne, S. (Ed.). 18(1), 15–18. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2542892/.
Falco, M. (2012). Whooping cough cases approaching highest levels in half a century. CNN. Retrieved from http://thechart.blogs.cnn.com/2012/07/19/whooping-cough-cases-approaching-highest-levels-in-half-a-century/?hpt=hp_t2.
Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison III, C. A., & Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 6(5), 343-347. Retrieved from http://www.nature.com/nmeth/journal/v6/n5/full/nmeth.1318.html.
Huth, J. R., Norton S. E., Shikone, Lockridge, O., Shikone, T., Hsueh, A. J., & Ruddon, R. W. (1994). Bacterial expression and in vitro folding of the beta-subunit of human chorionic gonadotropin (hCG beta) and functional assembly of recombinant hCG beta with hCG alpha. Endocrinology. 135(3), 911-918.
Lee, S. Y. (1996). High cell-density culture of Escherichia coli. Trends in Biotechnology. 14(3), 98-105. Retrieved from http://www.sciencedirect.com/science/article/pii/0167779996809309.
Lu, T.K., Collins, J.J. (2007). Dispersing biofilms with engineered enzymatic bacteriophage. PNAS 104(27):11197-11202.
Qimron, U., Tabor, S., & Richardson, C. C. (2010). New details about bacteriophage T7-host interactions. Microbe magazine. Retrieved from http://www.microbemagazine.org/index.php?option=com_content&view=article&id=1428:new-details-about-bacteriophage-t7-host-interactions&catid=376:featured&Itemid=452.