 "
"
Team:British Columbia/Pathway
From 2012.igem.org

Our Pathway Model
The study of environmental genomics attempts to capture the taxonomic and functional diversity of natural microbial communities. Our host at UBC, the Hallam lab, designs novel tools for analyzing the gene content in the context of distributed metabolism. Recently, a pipeline has been developed for the automated construction and visualizing of metabolic pathways from genomic data by integrating software such as Pathway Tools, Pathologic and Metacyc [1]. This provided us an opportunity to model pathway compartmentalization and distribution amongst microbes in the natural environment as it applies to our project.Summary of the pipeline for community-level metabolic analysis (Figure 1):
First, open reading frames are predicted from sequence data with Prodigal, are then annotated by protein BLAST, and later summarized in a GenBank file. Pathway/genome databases (PGDBs) are generated from sequence data in a manner which does not constrain predictions within the scope of model organisms. This produces a community-based analysis that can be visualized using Pathway Tools.
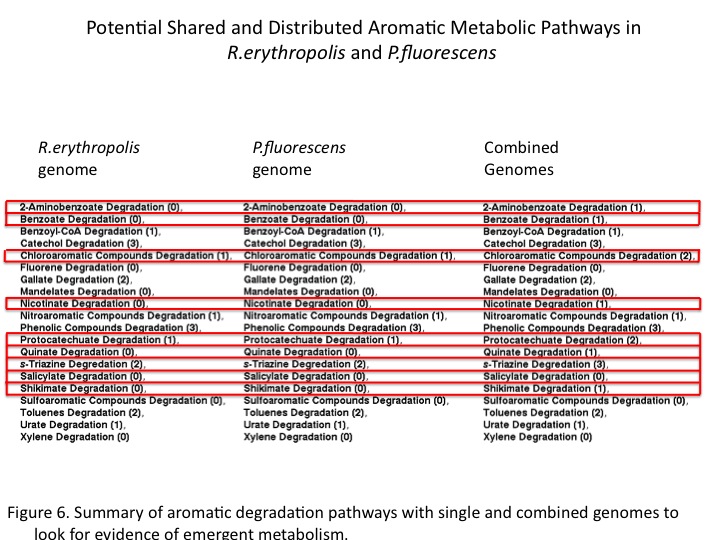
Modeling Metabolism:
Rhodococcus erythropolis and Pseudomnas fluorescens are often both prevalent in similar niches. With a high likelihood that these organisms encounter each other, studies have been conducted to assess their metabolic properties in co-culture. In a study by Kayser et al, it was shown that the 4S biodesulferization pathway in R. erythropolis demonstrated higher activity the presence of P. fluorescens [2]. As P. fluorescens does not biodesulferize DBT and sulfate is known to repress the 4S pathway, we looked to analyze the genomes of the two organisms in the context of sulfur metabolism. The 4S pathway releases sulfite, which is toxic to the cell, and therefore genomes were analyzed with the aforementioned pipeline for pathways involved in metabolizing sulfite. It was found that both organisms have annotated genes which convert sulfite into sulfate via a reductase, however the organisms differ in downstream metabolism of sulfate. While both organisms contain a pathway for assimilatory sulfate metabolism, only P. fluorescens has the capability for dissimilatory sulfate metabolism (Figure 2, 3). Based on these findings, we can hypothesize that R. erythropolis and P. fluorescens likely excrete and catabolize any excess sulfate. This provides an explanation for the improved desulfurization found in co-culture conditions. P. fluorescens potentially removes sulfate from the environment, allowing increased secretion by R. erythropolis, and thereby derepressing 4S pathway. The prediction of distributed sulfite metabolism to improve biodesulferization in the environment provides both a testable hypothesis, as well as a grounds for improving biodesulferization through synthetic pathway distribution.



Ultimately, gene annotation-based models for distributed metabolism in the environment may help to engineer and optimize complex metabolism through synthetic consortia.
References
[1] Hanson, N.W; Page, A.P; Konwar, K.M; Howes, C.G; Hallam, S.J. Metabolic interaction networks for the whole community, 2012. Unpublished. [2] Kayser, K.J; Biolaga-Jones, B.A.; Jackowski, K; Odusan, O; Kildane, J.J. Utilzation of organosulfur compounds by anexic and mixed culture of ''Rhodococcus rhodochrous'' IGTS8, 1993. Journal of General Microbioology, 139: 3123-3129. [3] Goswami, M; Shivaraman, N; Singh, R.P. Microbial metabolism of 2-chlorophenol, phenol and p-cresol by ''Rhodococcus erythropolis'' M1 and co-culture with ''Pseudomonas fluorescens'' P1, 2005. Microbiological Research, 160: 101-109.