"
"
Team:British Columbia/Pathway
From 2012.igem.org

Our Pathway Model
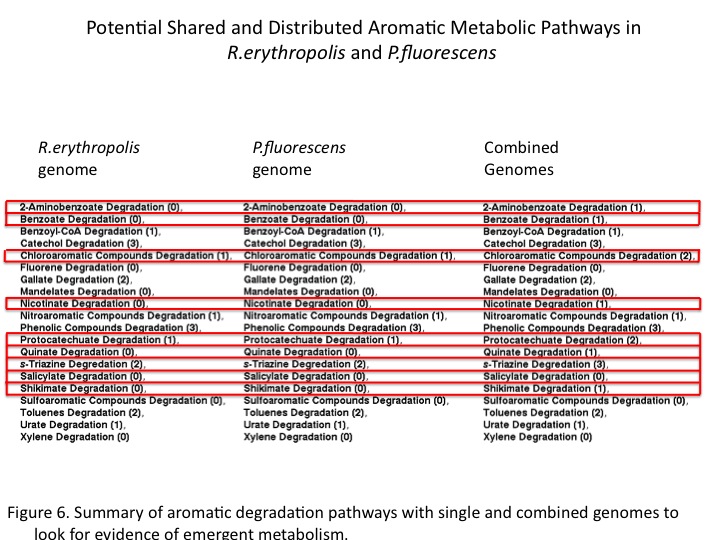
The study of environmental genomics attempts to capture the taxonomic and functional diversity of natural microbial communities. Our host at UBC, the Hallam lab, designs novel tools for analyzing the gene content in the context of distributed metabolism. Recently, a pipeline has been developed for the automated construction and visualizing of metabolic pathways from genomic data by integrating software such as Pathway Tools, Pathologic and Metacyc [1]. This provided an opportunity to model pathway compartmentalizing and distribution between microbes in the natural environment as it applies to our project. Summary of the pipeline for community-level metabolic analysis (Figure 1): First, open reading frames predicted from sequence data using Prodigal are annotated by protein BLAST and summarized in a GenBank file. Pathway/genome databases (PGDBs) are generated from sequence data in a manner that does not constrain predictions within the scope of model organisms. This allows for a community-based analysis that can be visualized using Pathway Tools.