 "
"
Team:Cambridge/Overview/Labbook
From 2012.igem.org
(→Week 3=) |
(→General Labbook) |
||
| Line 180: | Line 180: | ||
*Progress so far: (1) made 25ug/ml chloramphenicol plates; (2) Transformed E. coli and B. subtilis using the corresponding [[Team:Cambridge/Protocols| Protocols]] with the Vibrio LuxBrick from Cambridge 2010 ([http://partsregistry.org/Part:BBa_K325909 BBa_K325909]); (3) Plated the transformed cells and incubate at 37 degrees celsius overnight | *Progress so far: (1) made 25ug/ml chloramphenicol plates; (2) Transformed E. coli and B. subtilis using the corresponding [[Team:Cambridge/Protocols| Protocols]] with the Vibrio LuxBrick from Cambridge 2010 ([http://partsregistry.org/Part:BBa_K325909 BBa_K325909]); (3) Plated the transformed cells and incubate at 37 degrees celsius overnight | ||
*Nanodrop was used to determine the concentration of DNA in the biobrick after resuspension. The result reading was 103ng/ul. | *Nanodrop was used to determine the concentration of DNA in the biobrick after resuspension. The result reading was 103ng/ul. | ||
| + | |||
| + | |||
| + | ==Week 7== | ||
| + | ===Thursday (09/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/Chemicallycompetentcells|Making chemically competent e.coli]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | * [[Team:Cambridge/Protocols/SOB|SOB]] made up. | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/PCRProtocol|PCR of positive control fragments]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Gibson positive control fragments are amplified using PCR | ||
| + | |||
| + | :* sfGFP from sfGFP-ampR (~700bp) | ||
| + | |||
| + | :* pSB4K5 backbone which confers kanamycin resistance (~3.3kb) | ||
| + | |||
| + | *Results: successful amplification; sfGFP (lanes 1-3); pSB4K5 (lanes 4-6) | ||
| + | |||
| + | '''Ratiometrica-Lux: [[Team:Cambridge/Protocols/PCRcolony|PCR of Lux vector]] | ||
| + | |||
| + | ---- | ||
| + | |||
| + | * Split mOrange vector amplification attempted again. | ||
| + | |||
| + | *Results: no bands again. | ||
| + | |||
| + | ===Friday (10/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/Chemicallycompetentcells|Making chemically competent e. coli]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *[[Team:Cambridge/Protocols/GlycerolStocks|Glycerol stocks]] of TOP10 are made competent, aliquoted into 1.5mL eppendorfs in two batches and stored in SOB at -80°C | ||
| + | |||
| + | *50μl are transformed with pUC19 control and plated on LB agar plates to test for competence | ||
| + | |||
| + | ===Sunday (12/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/Gibsonassembly|Assembly of sfGFP construct from known functional DNA fragments]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Fragments provided by [[Team:Cambridge/Team|Fernan]] assembled with Gibson. | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/TransformationofE.coli|Transformation of e.coli with fluorescent construct and sfGFP positive control]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Chemically competent e.coli cells transformed with fluorescent construct Gibson product from 07/08/12. | ||
| + | |||
| + | *E.coli cells also transformed with sfGFP Gibson product made earlier today in triplicate. | ||
| + | |||
| + | *All transformants plated out on 100 μg/ml ampicillin. | ||
| + | |||
| + | *sfGFP fragments have produced successful Gibson products in the past, so this will act as our positive control. If cells grow with the plasmid and fluoresce properly, we will know the master mix works. Otherwise, we will remake the master mix. | ||
| + | |||
| + | *Results: there is growth on the ampicillin plates but very little- we suspect a problem with positive control fragments DNA concentration being too low. (13/8/12) | ||
| + | |||
| + | ==Week 8== | ||
| + | ===Tuesday (14/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/Gibsonassembly|Gibson assembly of positive control]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Fragments from [[Team:Cambridge/Lab_book/Week_7#Thursday_.2809.2F08.2F12.29|Tom's PCR]] from last week are used: the PSB4K5 backbone (in triplicates), and sfGFP from sfGFP-ampR (in triplicates) | ||
| + | |||
| + | *Protocol changed slightly: 0.5 μl of each DNA fragment solution mixed in with master mix to make up 4 μl total volume (1μl DNA and 3μl master mix). | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/TransformationofE.coli|Transformation of e.coli with positive control DNA]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Chemically competent e.coli cells transformed with plasmid DNA produced by Gibson assembly step. | ||
| + | |||
| + | *Transformants plated out on 50 μg/ml kanomycin plates. Incubated overnight at 37 °C. | ||
| + | |||
| + | ===Wednesday (15/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/PCRProtocol|PCR of positive control fragments]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *A replication of [[Team:Cambridge/Lab_book/Week_7#Thursday_.2809.2F08.2F12.29|last week's PCR]],except each fragment is done 5 times to generate more fragments so that the positive control could be used for future experiments | ||
| + | |||
| + | *Results: successful amplification of all fragments. | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/GelExtractionofDNA|Extraction of positive control DNA]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Positive control DNA from gel excised and purified. | ||
| + | |||
| + | *Additional elution steps used to concentrate resultant solution: eluants are recycled at least once into the spin column | ||
| + | |||
| + | *Verified with nanodropper - final DNA concentrations are around 20ng/ul for first eluants, and 15ng/ul for second eluants; this is twice as concentrated as before | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/Gibsonassembly|Gibson assembly of positive control DNA]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | * A combination of fragments from the PCR are chosen: the highest, second highest, third highest, lowest concentration first eluants, and the highest concentration second eluants are put together | ||
| + | |||
| + | * Reading that our T5 exonuclease concentration in Gibson assembly may be too high, we made up some mastermix with 1/5 of the T5 exo concentration and tested that in parallel with the fragments of various concentrations (2x5 = 10 Gibson reactions in total) | ||
| + | |||
| + | * Results: Successful Gibson assembly reactions. The lowered T5 exo conc seems to lower the Gibson efficiency, and the more concentrated fragments seem to produce more colonies. | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/TransformationofE.coli|Transformation of positive control DNA into e.coli]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Chemically competent e.coli transformed with positive control Gibson products made previously. | ||
| + | |||
| + | *Transformants plated out onto 50 μg/ml kanomycin plates and incubated at 37 °C overnight. | ||
| + | |||
| + | == Week 9 == | ||
| + | |||
| + | ===Tuesday (21/08/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/PCRProtocol|PCR of positive control]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Our order of Phusion has arrived- though we are still not confident with our PCR so we decided to first test with sfGFP | ||
| + | |||
| + | *Standard PCR conditions are used | ||
| + | |||
| + | *Results: success | ||
| + | |||
| + | == Week 10 == | ||
| + | '''Gibson efficiency diagnostics''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *We decided to try to get to the bottom of why our Gibson assembly is so inefficient. Until this is fixed, we won't be able to do anything higher than 2-part reactions. | ||
| + | |||
| + | *PJ, a member of the Haseloff lab whose gibson assemblies are working efficiently agreed to help us. Using our control fragments, PJ assembled them with our master mix and his master mix, and transformed them into his competent E.coli. Similarly, we assembled our fragments (from the same tube) with our master mix, but using his competent cells and our competent cells. This should show us if there is a problem with our mix or cells. | ||
| + | |||
| + | == Week 11== | ||
| + | ===Monday (03/09/12)=== | ||
| + | |||
| + | '''Further Gibson diagnostics''' | ||
| + | |||
| + | No difference between PJs cells and my cells, or PJs master mix and mine, but he still obtains > 10x higher efficiencies. | ||
| + | |||
| + | Tonight, we both assembled with my positive control again, but this time used our plates and his plates to plate the cells, to check for any difference there. | ||
| + | |||
| + | ===Tuesday (04/09/12)=== | ||
| + | |||
| + | '''Further Gibson Diagnostics''' | ||
| + | |||
| + | Considered it might be the PCR machine. Theirs is a hot-block, ours is a hot-air/centrifuge - type. This allows them to transfer reactions from ice to 50 C rapidly by preheating the block, theoretically making them much better able to limit the activity of the T5 exonuclease (which should only chew away a couple of dozen bases from the 3', ideally). | ||
| + | |||
| + | Direct comparison of their PCR machine and ours for another assembly with our positive control. | ||
| + | |||
| + | ===Wednesday (05/09/12)=== | ||
| + | '''Further Gibson diagnostics''' | ||
| + | |||
| + | Direct comparison between PCR machines showed a >10x difference in efficiency. We have isolated most of our efficiency problems to our PCR machine. | ||
| + | |||
| + | ==Week 12== | ||
| + | ===Monday (10/09/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/PCRProtocol|PCR of biobrick vector DNA]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Attempted to run PCR of the backbone for the biobricks once more. Settings: Annealing temperature: 56°C, elongation time: 30secs. | ||
| + | |||
| + | *After running on gel, saw that while the -8 vector amplification appears to be producing some correctly sized (~2kb) bands, the other two only seem to be forming many primer dimers. Analysis of the sequence of the prefix and suffix revealed a CG palindrome that appears to be causing self annealing. Inserts are not affected, as they have the palindrome at the 5' end of the primer. | ||
| + | |||
| + | *Will retry at higher temperatures tomorrow. | ||
| + | |||
| + | ===Tuesday (11/09/12)=== | ||
| + | |||
| + | '''[[Team:Cambridge/Protocols/PCRProtocol|PCR of biobrick backbone]]''' | ||
| + | |||
| + | ---- | ||
| + | |||
| + | *Retried PCR of backbone for +8 magnesium riboswitch biobrick and fluoride biobrick at 60 °C and 64 °C. | ||
| + | |||
| + | *Verification gel showed that no bands of the appropriate size came out. Once more, only primer dimers produced. | ||
| + | |||
| + | *Production of primer dimers at these temperatures causes us to doubt that a successful PCR will be carried out at these temperatures. We may have to switch to ordinary ligation to construct our biobricks for submission. | ||
Latest revision as of 02:36, 27 October 2012
Contents |
Judging Form
- Please help the judges by filling out this form. Tell them what medal you think you deserve and why. Tell them which special prizes you should win. Help them find your best parts. Show them how you thought about the safety of your project. Helping the judges will help you too.
- Team: Cambridge
- Region: Europe
- iGEM Year:2012
- Track:Foundational Advance
- Project Name:Parts for a reliable and field ready biosensing platform
- Project Abstract: Implementation of biosensors in real world situations has been made difficult by the unpredictable and non-quantified outputs of existing solutions, as well as a lack of appropriate storage, distribution and utilization systems. This leaves a large gap between a simple, functional sensing mechanism and a fully realised product that can be used in the field.
We aim to bridge this gap at all points by developing a standardised ratiometric luciferase output in a Bacillus chassis. This output can be linked up with prototyped instrumentation and software for obtaining reliable quantified results. Additionally, we have reduced the specialized requirements for the storage and distribution of our bacteria by using Bacillus' sporulation system. To improve the performance of our biosensing platform we have genetically modified Bacillus’ germination speed. Lastly, we demonstrated the robustness of our system by testing it with a new fluoride riboswitch, providing the opportunity to tackle real life problems.
iGEM Medals for non-software teams
- We believe our team deserves the following medal:
- Bronze
- Silver
- √Gold
Because we met the following criteria (check all that apply and provide details where needed)
Requirements for a Bronze Medal
- √Register the team, have a great summer, and plan to have fun at the Regional Jamboree.
- √Successfully complete and submit this iGEM 2012 Judging form.
- √Create and share a Description of the team's project using the iGEM wiki and the team's parts using the Registry of Standard Biological Parts.
- √Plan to present a Poster and Talk at the iGEM Jamboree.
- √Enter information detailing at least one new standard BioBrick Part or Device in the Registry of Standard Biological Parts. Including:
- √Primary nucleaic acid sequence
- √Description of function
- √Authorship
- Safety notes, if relevant.
- √Acknowedgment of sources and references
- √Submit DNA for at least one new BioBrick Part or Device to the Registry.
Additional Requirements for a Silver Medal
- √Demonstrate that at least one new BioBrick Part or Device of your own design and construction works as expected; characterize the operation of your new part/device.
- √Enter this information and other documentation on the part's 'Main Page' section of the Registry
Part Number(s): [http://partsregistry.org/Part:BBa_K911004 BBa_K911004]
Additional Requirements for a Gold Medal: (one OR more)
- Improve an existing BioBrick Part or Device and enter this information back on the Experience Page of the Registry.
Part Number(s): None - √Help another iGEM team by, for example, characterizing a part, debugging a construct, or modeling or simulating their system.
Link to this information on your wiki. Page name: Team:Cambridge/Outreach/Collaboration - √Outline and detail a new approach to an issue of Human Practice in synthetic biology as it relates to your project, such as safety, security, ethics, or ownership, sharing, and innovation.
Link to this information on your wiki.
Page name: Team:Cambridge/HumanPractices/Overview,Team:Cambridge/HumanPractices/MarketResearch,Team:Cambridge/HumanPractices/FutureDirections
iGEM Prizes
All teams are eligible for special prizes at the Jamborees. more... To help the judges, please indicate if you feel you should be evaluated for any of the following special prizes:
- √Best Human Practice Advance
- √Best Experimental Measurement
- Best Model
Please explain briefly why you should receive any of these special prizes:
Best Human Practice Advance:
We feel that we deserve this prize for three reasons:
- We explored the impacts, *both positive and negative*, of synthetic biology as a solution to real world problems, through interviewing professionals working in a relevant field, namely the impact of arsenic water contamination in Bangladesh.
- We recognized existing problems with the way the current direction of synthetic. On going through the registry we found that most of the characterization data for biosensing parts is often neither comparable nor replicable. We have worked to solve this issue, for example with our ratiometric dual channel output.
- *Our project doesn’t stop here*, in Chanel number 6 (Team:Cambridge/HumanPractices/FutureDirections) we considered the future implications and technological applications of our project, as well as the means by which it could be improved by subsequent users. We feel that the end to an iGEM project should not be the conclusion of an idea, but the start of it.
Best BioBrick Measurement Approach:
It is absolutely vital that a quantitative, numerical, robust, and flexible measurement approach exists to relay information to a user that is an accurate representation of the input processed by a biological device. Working from these principles, the following was done:
- We designed and built Biologger, a *cheap, arduino-based, fully functional automatic rotary device* that has an incorporated ratiolumnometer
- Our project is entirely open-sourced and open-platform. We have published source code for the two applications which serve to operate the device, one for PCs and the other for Android devices, as well as the open source circuit design that provides this ratiometric reading. Furthermore, the Android app is able to receive its data wirelessly, which we feel is a great advance in BioBrick measurement.
- Our dual-channel luciferase reporter was successfully tested with a dilution series of E.coli transformed with the Lux Operon (under pBAD) biobrick (Part BBa_K325909) of the Cambridge iGEM 2010 team. It can detect, with good accuracy, both different light intensities, as well as the percentages of blue or orange frequencies in a sample.
- Our device was successfully tested using artificial light to detect different frequencies (colours) as well.
Having done all the above, we believe that this fully open-sourced instrumentation kit (mechanical) chassis, electronics, software code), estimated at *$35.00* (or $85.00 if a Bluetooth modem is required), is a complete BioBrick measurement solution for any and all BioBricks with a light output.
Team_Parts
To help the judges evaluate your parts, please identify 3 of your parts that you feel are best documented and are of the highest quality.
- Best new BioBrick part (natural)
- [http://partsregistry.org/Part:BBa_K911003 BBa_K911003]
- Best new BioBrick part (engineered)
- [http://partsregistry.org/Part:BBa_K911004 BBa_K911004]
- Best improved part(s): None
List any other parts you would like the judges to examine:[http://partsregistry.org/Part:BBa_K911001 BBa_K911001], [http://partsregistry.org/Part:BBa_K911008 BBa_K911009], [http://partsregistry.org/Part:BBa_K911008 BBa_K911008]
Please explain briefly why the judges should examine these other parts:
- Magnesium Sensitive Riboswitch [http://partsregistry.org/Part:BBa_K911001 BBa_K911001]
As a riboswitch sensing construct, this part is an entirely new type of biosensor (along with the fluoride construct) that could potentially change the way we think about designing input genetic circuits. Unlike the fluoride riboswitch, it is a derepression system and therefore serves to demonstrate the principle that riboswitches can be used regardless of whether they turn on or off their reporter. - Fluorescent ratiometric construct for standardizing promoter output [http://partsregistry.org/Part:BBa_K911009 BBa_K911009]
Fluorescence is a major cornerstone for biosensors in the registry, however, most parts do not involve the use of a ratiometric output, which has been shown in the literature to provide much more reliable and meaningful data. This part not only furthers the development of ratiometric measurements in molecular biology but due to the choice of promoters and terminators it can be used to characterize the difference in activity between E. coli and B. Subtilis - Fast Germination (B. subtilis) [http://partsregistry.org/Part:BBa_K911008 BBa_K911008]
This part is entirely novel for the registry and fully utilizes the recombination machinery inherent in the Bacillus chassis. Have spores that can germinate at a faster rate is certainly a worthy achievement and could help with experiments with B. Subtilis that any future iGEM teams may wish to perform.
iGEM Safety
For iGEM 2012 teams are asked to detail how they approached any issues of biological safety associated with their projects.
The iGEM judges expect that you have answered the four safety questions Safety page on your iGEM 2012 wiki.
Please provide the link to that page: Page name: Team:Cambridge/Safety
Attribution and Contributions
For iGEM 2012 the description of each project must clearly attribute work done by the team and distinguish it from work done by others, including the host labs, advisors, and instructors.
Please provide the link to that page, or comments in the box below: Page name: Team:Cambridge/Attributions
Comments
If there is any other information about your project you would like to highlight for the judges, please provide a link to your wiki page here: Team:Cambridge/Overview/DesignProcess
General Labbook
Week 3
Monday (09/07/12)
- Bacillus salts *10 made up and autoclaved.
- Medium A base *10 made up (glucose will be added tomorrow)
- bacillus strain 168 streaked out and grown on plate overnight.
Tuesday (10/07/12)
- Filtered glucose added to sterile Medium A base.
- Sterile aliquots of salts and medium apportioned and put in fridge (10*50ml each)
- Needed to make up MgCl2 and CaCl2 solutions of the correct concentrations. Required multiple dilution steps:
- MgCl2 (250nM), Mr = 147 Da, make 100ml of solution
- 0.508g of MgCl2 dissolved in 100ml of H2O
- 1ml of this solution mixed with 99ml of H2O
- 0.1ml of this solution mixed with 99.9ml of H2O
- CaCl2 (50mM), Mr = 203.3 Da, make 100ml of solution
- 0.735g of CaCl2 dissolved in 100ml of H2O
- Made up Medium A (150ml) and Medium B (50ml)
Wednesday (11/07/12)
- Colonies of strain 168 removed from plate and added to three separate conical flasks, each with 48ml of Medium A inside. OD650 readings taken until enough cells were added such that absorbance was between 0.1 and 0.2.
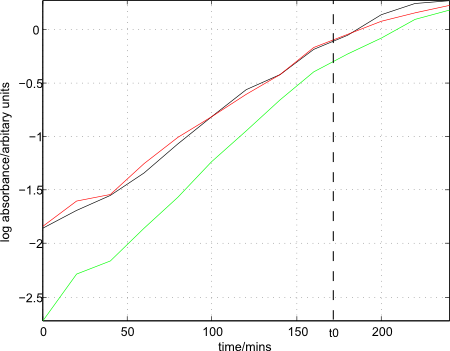 bacillus growth curves
bacillus growth curves

- Flasks placed inside a shaking incubator (200rpm) and samples taken every 20mins until growth had leveled off, as determined by the concurrently plotted growth curves.
- Growth curves, as well as t0 (at 170mins) plotted:
- 90 mins after t0 (at 260 mins), cells removed from shaking incubator.
- 0.45ml of Medium B pre-warmed in Eppendorf tubes.
- 20 individual samples of 0.05ml taken from each growth flask and added to Eppendorfs.
- Eppendorfs placed back in shaking incubator for 60 mins with lids off.
- Eppendorfs centrifuged at ~13000 rpm for 10 mins, supernatant removed.
- 60% glycerol added (0.5ml/tube) and tubes vortexed.
- All 60 tubes now frozen at -80 °C.
- Edited protocol so that it actually works.

Arduino circuitry
- Real-Time image captured by our freshly made primitive software, monitoring a light sensor on an arduino board. C++ was used to communicate with the Arduino and Python was used for data logging.
Thursday (12/07/12)
Test transformation of frozen bacillus stocks
- Two samples of bacillus defrosted from different batches.
- Supernatant spun off, bacterial pellet isolated and 0.1ml of Medium B added.
- Plasmid Tag RFP-T unfrozen. Concentration of DNA = 360ng/μl, so 2μl needed for the desired 0.6μg of DNA. DNA added to bacteria.
- Eppendorfs placed in shaking incubator at 30 °C and 180rpm for 60 mins.
- Bacteria plated on choramphenicol containing plates. Aseptic technique was used as far as possible.
Friday (13/07/12)
Test transformation of bacillus stocks

- Results: In this image, several colonies have clearly gained pink coloration from the transfected plasmid. This demonstrates that our stocks should be usable. However, the large number of opportunistic colonies that do not appear to have been transfected means that we will have to be careful to check that our transfected bacteria contain what is expected.
Transformation of bacillus and e.coli with lux genes from 2010
- Progress so far: (1) made 25ug/ml chloramphenicol plates; (2) Transformed E. coli and B. subtilis using the corresponding Protocols with the Vibrio LuxBrick from Cambridge 2010 ([http://partsregistry.org/Part:BBa_K325909 BBa_K325909]); (3) Plated the transformed cells and incubate at 37 degrees celsius overnight
- Nanodrop was used to determine the concentration of DNA in the biobrick after resuspension. The result reading was 103ng/ul.
Week 7
Thursday (09/08/12)
Making chemically competent e.coli
- SOB made up.
PCR of positive control fragments
- Gibson positive control fragments are amplified using PCR
- sfGFP from sfGFP-ampR (~700bp)
- pSB4K5 backbone which confers kanamycin resistance (~3.3kb)
- Results: successful amplification; sfGFP (lanes 1-3); pSB4K5 (lanes 4-6)
Ratiometrica-Lux: PCR of Lux vector
- Split mOrange vector amplification attempted again.
- Results: no bands again.
Friday (10/08/12)
Making chemically competent e. coli
- Glycerol stocks of TOP10 are made competent, aliquoted into 1.5mL eppendorfs in two batches and stored in SOB at -80°C
- 50μl are transformed with pUC19 control and plated on LB agar plates to test for competence
Sunday (12/08/12)
Assembly of sfGFP construct from known functional DNA fragments
- Fragments provided by Fernan assembled with Gibson.
Transformation of e.coli with fluorescent construct and sfGFP positive control
- Chemically competent e.coli cells transformed with fluorescent construct Gibson product from 07/08/12.
- E.coli cells also transformed with sfGFP Gibson product made earlier today in triplicate.
- All transformants plated out on 100 μg/ml ampicillin.
- sfGFP fragments have produced successful Gibson products in the past, so this will act as our positive control. If cells grow with the plasmid and fluoresce properly, we will know the master mix works. Otherwise, we will remake the master mix.
- Results: there is growth on the ampicillin plates but very little- we suspect a problem with positive control fragments DNA concentration being too low. (13/8/12)
Week 8
Tuesday (14/08/12)
Gibson assembly of positive control
- Fragments from Tom's PCR from last week are used: the PSB4K5 backbone (in triplicates), and sfGFP from sfGFP-ampR (in triplicates)
- Protocol changed slightly: 0.5 μl of each DNA fragment solution mixed in with master mix to make up 4 μl total volume (1μl DNA and 3μl master mix).
Transformation of e.coli with positive control DNA
- Chemically competent e.coli cells transformed with plasmid DNA produced by Gibson assembly step.
- Transformants plated out on 50 μg/ml kanomycin plates. Incubated overnight at 37 °C.
Wednesday (15/08/12)
PCR of positive control fragments
- A replication of last week's PCR,except each fragment is done 5 times to generate more fragments so that the positive control could be used for future experiments
- Results: successful amplification of all fragments.
Extraction of positive control DNA
- Positive control DNA from gel excised and purified.
- Additional elution steps used to concentrate resultant solution: eluants are recycled at least once into the spin column
- Verified with nanodropper - final DNA concentrations are around 20ng/ul for first eluants, and 15ng/ul for second eluants; this is twice as concentrated as before
Gibson assembly of positive control DNA
- A combination of fragments from the PCR are chosen: the highest, second highest, third highest, lowest concentration first eluants, and the highest concentration second eluants are put together
- Reading that our T5 exonuclease concentration in Gibson assembly may be too high, we made up some mastermix with 1/5 of the T5 exo concentration and tested that in parallel with the fragments of various concentrations (2x5 = 10 Gibson reactions in total)
- Results: Successful Gibson assembly reactions. The lowered T5 exo conc seems to lower the Gibson efficiency, and the more concentrated fragments seem to produce more colonies.
Transformation of positive control DNA into e.coli
- Chemically competent e.coli transformed with positive control Gibson products made previously.
- Transformants plated out onto 50 μg/ml kanomycin plates and incubated at 37 °C overnight.
Week 9
Tuesday (21/08/12)
- Our order of Phusion has arrived- though we are still not confident with our PCR so we decided to first test with sfGFP
- Standard PCR conditions are used
- Results: success
Week 10
Gibson efficiency diagnostics
- We decided to try to get to the bottom of why our Gibson assembly is so inefficient. Until this is fixed, we won't be able to do anything higher than 2-part reactions.
- PJ, a member of the Haseloff lab whose gibson assemblies are working efficiently agreed to help us. Using our control fragments, PJ assembled them with our master mix and his master mix, and transformed them into his competent E.coli. Similarly, we assembled our fragments (from the same tube) with our master mix, but using his competent cells and our competent cells. This should show us if there is a problem with our mix or cells.
Week 11
Monday (03/09/12)
Further Gibson diagnostics
No difference between PJs cells and my cells, or PJs master mix and mine, but he still obtains > 10x higher efficiencies.
Tonight, we both assembled with my positive control again, but this time used our plates and his plates to plate the cells, to check for any difference there.
Tuesday (04/09/12)
Further Gibson Diagnostics
Considered it might be the PCR machine. Theirs is a hot-block, ours is a hot-air/centrifuge - type. This allows them to transfer reactions from ice to 50 C rapidly by preheating the block, theoretically making them much better able to limit the activity of the T5 exonuclease (which should only chew away a couple of dozen bases from the 3', ideally).
Direct comparison of their PCR machine and ours for another assembly with our positive control.
Wednesday (05/09/12)
Further Gibson diagnostics
Direct comparison between PCR machines showed a >10x difference in efficiency. We have isolated most of our efficiency problems to our PCR machine.
Week 12
Monday (10/09/12)
- Attempted to run PCR of the backbone for the biobricks once more. Settings: Annealing temperature: 56°C, elongation time: 30secs.
- After running on gel, saw that while the -8 vector amplification appears to be producing some correctly sized (~2kb) bands, the other two only seem to be forming many primer dimers. Analysis of the sequence of the prefix and suffix revealed a CG palindrome that appears to be causing self annealing. Inserts are not affected, as they have the palindrome at the 5' end of the primer.
- Will retry at higher temperatures tomorrow.
Tuesday (11/09/12)
- Retried PCR of backbone for +8 magnesium riboswitch biobrick and fluoride biobrick at 60 °C and 64 °C.
- Verification gel showed that no bands of the appropriate size came out. Once more, only primer dimers produced.
- Production of primer dimers at these temperatures causes us to doubt that a successful PCR will be carried out at these temperatures. We may have to switch to ordinary ligation to construct our biobricks for submission.