 "
"
Team:Valencia/genetic engineering
From 2012.igem.org
Genetic Engineering

Cyanobacteria are great organisms to be used in Synthetic Biology because of their ability to capture solar energy and CO2 and the fact that they can be easily genetically manipulated due to its small genome and their capacity to accept foreign DNA naturally. For our project we have chosen the cyanobacteria Synechococcus elongatus (figure 1) for genetic engineering because is the model organism for studying some prokaryotic processes (there is a lot of information of how to transform it) and in the last years it has become a model organism for some industrial processes, like biofuel production (Wang et al. 2012).
S. elongatus has a circular genome of ≈2.7Mb ( fully sequenced ) with a GC content of 55.5%, which contains the genes for 2.612 proteins and 53 RNAs (Atsumi et al. 2009).
Transforming S. elongatus PCC7942 for promoter characterization:
Our main objective has been to characterize the psbAI promoter (Submitted parts) of Synechococcus elongatus PCC7942 in order to know more about its operation and understand how this promoter would control our final construct ( Designed parts) to have a diel switch of AHL, the signal molecule for our Aliivibrio fischeri population to glow.
To characterize this promoter we had two options: making psbAIp::lacZ fusions and monitored the β-galactosidase activity or making psbAIp::luxABCDE fusions and monitored the bioluminescence produced as a result of the promoter activation. We refused to use a psbAIp::lacZ fusion because some assays report that the psbAI promoter response to light cannot be properly monitored by the β-galactosidase activity (Nair et al. 2001). For this reason we choosed using vector fusions. With this aim, we cloned several fusion plasmids (pAM977 and pAM2195, table 1) in Escherichia coli and transformed them into S. elongatus, both wildtype and cscB strain. The fusion vectors were provided by Susan Golden´s lab.

Cloning into E. coli
We successfully cloned all the vectors in DH5α E. coli strains. For E. coli transformation we used this protocol. For the selective selection for pAM977 we used ampicillin (1μl/ml) and for pAM2195 (figure 2) Chloramphenicol (0.7μl/ml). The lack of spectrophotometer or other apparatus to measure optical density in our lab impede us to determine the concentration of our vectors.

Transforming Coccus
Once cloned we transformed a wild type strain provided from the Physiology, Genetic and Microbiology departments of University of Alicante and the cscB strain provided from the Wyss Institute in Harvard. As the cscB strain from Harvard was resistant to Cm, we only could transform it with the pAM977 plasmid, which gives Sp resistance. Wildtype was transformed with both plasmids (pAM977 and pAM2195).
We used the following protocol to transform Synechococcus:
Protocol
Extracted from Clerico et al. 2007:
- We grew 100ml of cscB and WT in BG-11 liquid medium shaking at 250rpm with constant light we used cold light fluorescent tubes. Cells had an optical density at 750nm (OD750) of 0.7, which is reach in 4 to 7 days approximately.
- We collected 15mL of the culture and centrifuge it at 6000g for 10 min and discarded the supernatant.
- We resuspended collected cells in 10 mL of 10mM NaCl and centrifuge them again at 6000g for 10 min.
- After it, we resuspended the pellet in 0.3mL of BG-11M liquid medium.
- We added 2μl of pAM2195 and pAM977 after doing a Miniprep of the clonation with a JetQuick kit
- Wrap tubes in aluminum foil to keep out light and incubate at 30oC for 15 to 20h with gentle agitation.
- And then, plate the entire 0.3mL cell suspension on BG-11 medium agar with the selective antibiotics. We used this concentration: 2μg/ml spectinomycin and 7.5μg/mL of chloramphenicol. After 7 to 10 days of incubation under standard light conditions transformed colonies should appear.
- After colonies appearance pick single transformants and grow them on fresh liquid BG-11M agar plate with selective antibiotic (this is done to ensure that all colonies have incorporated the trans gene as S. elongatus has multiple copies of its chromosome).
- After 5 to 7 days of growth, cyanobacteria can be used to inoculate a BG-11 liquid culture with the selective antibiotic.
Results
We weren´t able to obtain transformants with the wild type strain of S. elongatus. But we obtained resistant colonies to Sp of the cscB strain transformed with the pAM977 vector (figure 4). This was achieved the last week of lab work, so due to the slowly growth rate of S. elongatus, we haven´t have the time to grow the transformants in liquid medium and start with the bioluminescence measures to characterize the promoter. However, we will try to have some results for the Jamboree concerning this part.
Transformation of E. coli for AHL production to test bioluminescent response in A. fischeri:
Another part of our project included transforming E. coli with the registry part BBa_K084014 in order to make E. coli produce AHL under IPTG regulation to test the bioluminescent response in A. fischeri. To achieve this goal we cloned the part in DH5α E. coli strains using this protocol(figure 5). The part (869 bp) is included inside the pSB1A2 (2079bp) BioBrick.
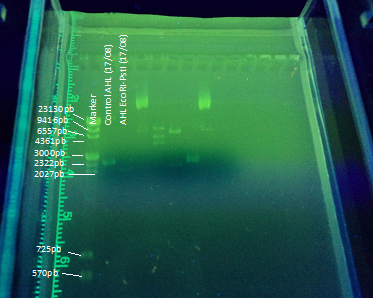
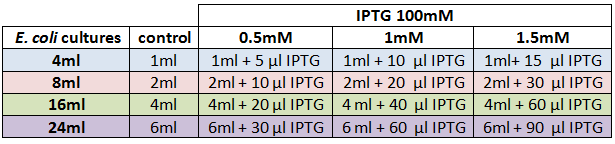
We designed an experimental procedure to test different experimental conditions for the production of AHL.
When we wanted to test if A. fischeri was able to glow with the AHL produced by E. coli we discovered that the strain we had been growing in the lab was not V. fischeri, but V.mediterrani (GREAT!).
We will try to obtain V. fischeri as soon as possible to test this.
Building our BioBricks
psbAI BioBrick
Our promoter with the aproppiate suffix and preffix (289bp) was synthesized by Genscript and cloned into a puc57-Kan (2579bp) vector. We successfully digested the part and ligated it into the psb1C3 BioBrick from the parts registry (figure 6, 7 and 8). For this aim we followed the openwetware protocols, which you can find here.
Complete Construct
Our final objective is to have a S. elongatus capable to produce AHL during the night. For this we designed this new BioBrick controled by the psbAI promoter which is active in normal light conditions. Here you will find all the information concerning the different parts of the BioBrick.
We successfully cloned all the parts in DH5α E. coli strains using this protocol. We used the BioBrick Assembly Manual protocol to ligate our parts, but only were able to ligate the psbAI promoter to psb1C3.



Atsumi, S., Higashide, W., and Liao, J. C. (2009) Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol. 27:1177-1180
Clerico, E. M., Ditty, J. L. & Golden, S.S. (2007) Specialized Techniques for Site-Directed Mutagenesis in Cyanobacteria. Methods in Molecular Biology. 362:153–172.
Nair, U., Thomas, C. & Golden, S. S. (2001) Functional Elements of the Strong psbAI Promoter of Synechococcus elongatus PCC 7942. J. Bacteriology, 183:1740–1747.
Wang, B., Wang J., Zhang, W. & Meldrum, D. R. (2012) Application of Synthetic Biology in cyanobacteria and algae. Frontiers in Microbiology, doi: 10.3389/fmicb.2012.00344