From 2012.igem.org
(Difference between revisions)
|
|
| Line 11: |
Line 11: |
| | |} | | |} |
| | | | |
| - | ==Modeling the evolution of a population during MAGE==
| |
| | | | |
| - | ===Predicting the distribution of mutations===
| |
| | | | |
| - | Modeling mutations across evolving populations | + | ==Modeling the evolution of a population during MAGE== |
| - | Replacement efficiency calculation. For each ssDNA oligo produced by CAD-GENOME, the allelic replacement efficiency in E. coli is estimated using empirical relationships previously published in the documentation for optMAGE [5]. CAD-GENOME tabulates these predicted efficiencies in the ssDNA oligo output files and uses these efficiencies values to guide the user to the number of MAGE cycles necessary to create a population of cells including all of the desired set of mutations, as described below.
| + | The distribution of specific mutations in MAGE is a stochastic process that we model as as a function of each oligo's predicted efficiency of allelic replacement (which can be estimated in ''E. coli'' as discussed in "Programming cells by multiplex genome engineering and accelerated evolution," Wang & Isaacs et al 2009), assuming that each mutation event is binary and exclusive. Then a population after ''c'' cycles is a weighted sum of ''n'' Bernoulli trials, each zero if the oligo does not mutate its target 'i' and otherwise equal to the number ''r'' of mutations it induces. Given efficiencies of allelic replacement ''p'', this probability mass function becomes: |
| - | Estimated distribution of targeted mutations
| + | |
| - | The distribution of specific mutations in a population over cycles of multiplexed recombination is naturally a stochastic process controlled by diverse physiological effects and binding interactions. CAD-GENOME models this process as a function of each oligo's predicted efficiency of allelic replacement, assuming that each recombination event is always either complete or insignificant and that no two oligos compete for binding at the same site. This model returns the relative prevalence of each replacement, of each possible number of replacements per cell, of each possible minimum number of replacements per cell, the mean number of mutations per cell, and the variance of the same. These estimates will guide users seeking to guarantee, for instance, that their experiment will mutate some fraction of their specimens to carry all desired replacements before subjecting them to some selection pressure. | + | |
| | | | |
| - | Survey for off-target binding sites
| + | [[Media:Eqns1.png]] |
| - | Off-target binding could undercut our assumption that oligos do not compete or lead to other confounding phenotypes, so as a warning to the user of such exceptions, CAD-GENOME searches the specimen genome for regions with four base pairs or more of homology with any oligo, records each region of potential hybridization, then tabulates those regions' positions and sequences with an estimated free energy of hybridization and plots a log-scaled histogram of the same.
| + | [[Media:Eqns2.png]] |
| | | | |
| - | ===Implementation===
| + | In doing this, we have derived a more general form of the binomial distribution. Computing this PMF involves solving the subset sum problem, but we optimized our algorithm to avoid slowdowns by using a recursive formula (Wadyicki, Shah et al. 1973) for the occasional, simpler case when all oligos carry the same number of mutations, and in other cases a branched, dynammic programming algorithm (Horowitz and Sahni 1974). |
| - | Single-stranded DNA hybridization and minimum folding free energies are calculated using the UNAFold software package. This allows for the creation of optimized ssDNA oligos for optimal efficiency in producing desired mutants, and for estimating the significance of off-target binding sites. The BLAST+ alignment package is used for providing users with the top alignments for oligos to confirm that the expected locus is being targeted correctly [16]. In addition, BLAST+ is used for finding possible alternative hybridization events which may lower the efficiency of MAGE experiments.
| + | |
| | | | |
| - | Given our assumptions, we calculate the distribution of mutations in a population after $c$ cycles of multiplexed recombination as a weighted sum of $n$ independent Bernoulli trials $r_iX_i$, each equal to $0$ if an oligo does not replace the sequence at its targeted position $i$ and equal to $r_i$ if it does, thus inducing its $r_i$ encoded mutations. Given an accurate array of estimated efficiencies of allelic replacement $p_i$, then, we find the probability mass function <math> \mathrm{Pr}(K=k; c) </math>
| |
| | | | |
| - | as follows from elementary probability theory and as derived from generating functions in S1. The sum in (1) is over the set $D_k$ of all sets of oligos $A$ which carry a total of $k$ mutations; $\bar{A}$ is the set of oligos in the pool but not in $A$. If each oligo carries a single replacement with the same efficiency of allelic replacement, then (1) can condense to form the binomial distribution.
| + | ==Survey for off-target binding sites== |
| | + | Not all MAGE-induced mutations will be at the intended sites; to identify likely unintended mutations, we scripted a search of the genome using BLAST+ to find subsequences with four base pairs or more matching oligos in the MAGE oligo pool, and estimates the change in Gibbs energy likely upon hybridization at each such off-target pairing, using the UNAFold software package. |
| | | | |
| - | Computing the probability mass function of $K(c)$ can be intensive, as enumerating the sets in $D_k$ is equivalent to solving the NP-complete subset sum problem, but we speed the process in two ways: when $r=1$, by using a formula recursive in $k$ taking $O(c \cdot \mathrm{max}(k))$(Wadyicki, Shah et al. 1973), and otherwise, a branched, dynammic programming algorithm taking $O(c \cdot \mathrm{max}(2^{n/2}, n|D_k|))$ (Horowitz and Sahni 1974). The complementary cumulative distribution function of $K$ (that is, $\mathrm{Pr}(K \ge k; c)$) is then determined recursively in $O(c \cdot \mathrm{max}(k))$. To find the mean and variance of $K(c)$, we simply find the sums of the means and variances of the definitely independent terms $M_i(c)$ constituting $K(c)$. These can be determined from their generating functions $h_{M_i}$,
| + | Both of these scripts will be bundled into an cloud-based tool for genomic engineering (unpublished work). |
| | | | |
| - | ===Detecting of off-target binding sites===
| + | <gallery> |
| | + | Image:MAGEfig1.png |
| | + | Image:MAGEfig2.png |
| | + | Image:MAGEfig3.png |
| | + | Image:MAGEhistogram.png |
| | + | </gallery> |
Revision as of 03:25, 4 October 2012
Modeling the evolution of a population during MAGE
The distribution of specific mutations in MAGE is a stochastic process that we model as as a function of each oligo's predicted efficiency of allelic replacement (which can be estimated in E. coli as discussed in "Programming cells by multiplex genome engineering and accelerated evolution," Wang & Isaacs et al 2009), assuming that each mutation event is binary and exclusive. Then a population after c cycles is a weighted sum of n Bernoulli trials, each zero if the oligo does not mutate its target 'i' and otherwise equal to the number r of mutations it induces. Given efficiencies of allelic replacement p, this probability mass function becomes:
Media:Eqns1.png
Media:Eqns2.png
In doing this, we have derived a more general form of the binomial distribution. Computing this PMF involves solving the subset sum problem, but we optimized our algorithm to avoid slowdowns by using a recursive formula (Wadyicki, Shah et al. 1973) for the occasional, simpler case when all oligos carry the same number of mutations, and in other cases a branched, dynammic programming algorithm (Horowitz and Sahni 1974).
Survey for off-target binding sites
Not all MAGE-induced mutations will be at the intended sites; to identify likely unintended mutations, we scripted a search of the genome using BLAST+ to find subsequences with four base pairs or more matching oligos in the MAGE oligo pool, and estimates the change in Gibbs energy likely upon hybridization at each such off-target pairing, using the UNAFold software package.
Both of these scripts will be bundled into an cloud-based tool for genomic engineering (unpublished work).
 "
"
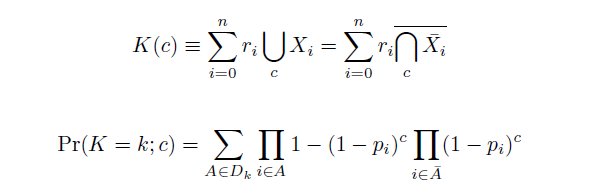{kind=link}
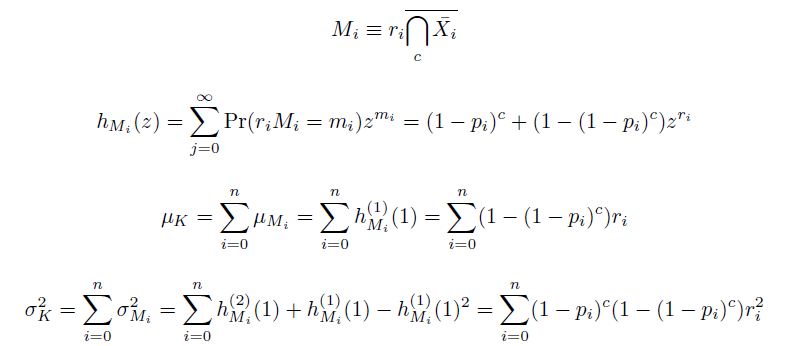{kind=link}



